

Summary
Contaminazione durante il sequenziamento genomico di organismi microscopici rimane un grande problema. Qui, vi mostriamo un metodo per sequenziare il genoma di un Tardigrada da un singolo esemplare con appena 50 pg di DNA genomic senza amplificazione intero genoma per ridurre al minimo il rischio di contaminazione.
Abstract
I tardigradi sono animali microscopici che entri in uno stato di ametabola anhydrobiosis chiamato fronte di essiccazione e può tornare al loro stato originale quando l'acqua è fornita. Il sequenziamento genomico di microscopici animali come i tardigradi rischi la contaminazione batterica che a volte porta a interpretazioni errate, ad esempio, per quanto riguarda il limite di trasferimento genico orizzontale in questi animali. Qui, forniamo un metodo di input ultrabasso per sequenziare il genoma di Tardigrada, Hypsibius dujardini, da un singolo esemplare. Impiegando rigorosa esclusione di lavaggio e contaminante insieme ad un'efficiente estrazione del 50 ~ 200 pg DNA genomico da un singolo individuo, abbiamo costruito una biblioteca sequenziata con uno strumento di sequenziamento del DNA. Queste librerie sono state altamente riproducibile e imparziale, e un'analisi informatica della sequenza si legge con altri genomi di H. dujardini ha mostrato una quantità minima di contaminazione. Questo metodo può essere applicato per i tardigradi saggiati che potrebbero non essere sequenziati utilizzando metodi precedenti.
Introduction
I tardigradi sono animali microscopici che possono entrare in uno stato di ametabola chiamato anhydrobiosis quando essiccazione di fronte. Recuperano dall'assorbimento di acqua1,2. Nello stato ametabola, i tardigradi sono in grado di tollerare i vari ambienti estremi, quali temperature estreme pressioni e3 4,5, un alto dosaggio di luce ultravioletta6, raggi x e raggi gamma 7 , 8e9di spazio cosmico. Dati genomici sono un fondamento indispensabile per lo studio dei meccanismi molecolari della anhydrobiosis.
Precedenti tentativi di sequenziare il genoma di tardigradi hanno mostrato segni di contaminazione batterica10,11,12,13,14. Sequenziamento genomico da tali piccoli organismi richiede un gran numero di animali ed è soggetto a contaminazione batterica; di conseguenza, abbiamo precedentemente stabilito un protocollo di sequenziamento utilizzando un metodo di input ultrabasso a partire da un singolo esemplare di Tardigrada, per ridurre al minimo il rischio di contaminazioni15. Utilizzando questi dati, abbiamo condotto ulteriormente una nuova sequenza di alta qualità e riassemblaggio del genoma di H. dujardini16,17. Qui descriviamo in dettaglio questo metodo di sequenziamento genomico da un singolo individuo tardigrade (nella figura 1). La convalida di questo metodo di sequenziamento è oltre il focus di questo lavoro e già è stata accuratamente esaminata nella nostra precedente relazione16.
Questo metodo è composto di due parti: l'isolamento di un singolo Tardigrada con il più basso possibile di contaminazione, l'estrazione di alta qualità dei livelli di pittogramma del DNA. Il Tardigrada è affamato e sciacquare accuratamente con acqua, così come gli antibiotici e osservato sotto un microscopio con ingrandimento X 500 per garantire la rimozione di qualsiasi contaminazione batterica. Misurazioni e stime precedenti indicano che un singolo individuo di Tardigrada contiene circa il 50-200 pg di genomic DNA16, che viene estratta per cracking l'esoscheletro di chitina dai cicli di gelo-disgelo o di omogeneizzazione manuale. Il DNA di genomic è presentato alla costruzione della libreria e sequenziato su uno strumento di sequenziamento del DNA. Un'analisi informatica ulteriori Mostra sequenziamento di alta qualità, così come i bassi livelli di contaminazione rispetto ai precedenti progetti di sequenziamento tardigrade.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. preparazione
- Preparare il gel di agarosio al 2% utilizzando acqua distillata (DW) come il solvente contenuto in una piastra di coltura plastica 90 mm e 10 mL di una cocktail con DW penicillina/streptomicina 1%. Il gel può essere conservato per 2-3 settimane in un incubatore a 18 ° C.
Nota: Evitare qualsiasi deposito del gel sotto i 10 ° C, per la bassa temperatura si ridurrà il gel di agarosio, risultante in un minuscolo gap tra il gel e la parete del piatto di cultura in cui i tardigradi possono essere intrappolati.
2. campione preparazione ed esclusione delle sostanze contaminanti
- Raccogliere un singolo Tardigrada, posizionarlo sulla piastra agar preparata e lavarlo 2x - 3x con DW per rimuovere eventuali particelle rimanenti.
- Incubare il Tardigrada a 18-22 ° C per 24 h per rimuovere qualsiasi cibo in eccesso dagli intestini.
- Posizionare il Tardigrada affamato in antibiotici penicillina/streptomicina per 2-6 h rimuovere qualsiasi contaminazione batterica e mettere l'animale decontaminato in un bicchiere pulito diapositiva utilizzando una pipetta P10.
- Osservare il Tardigrada sotto un microscopio a 500 ingrandimenti e confermare che non esistono nessun batteri rimanente.
Nota: L'alto ingrandimento con un microscopio stereoscopico è ottima, ma un microscopio ottico può essere utilizzato in alternativa senza applicare un vetrino coprioggetti. - Raccogliere l'individuo utilizzando una pipetta P10 con un massimo di 5 µ l di liquido, metterlo in una provetta di reazione a catena (PCR) di basso-associazione della polimerasi e rimuovere come liquido molto in eccesso possibile.
Nota: Le microprovette, provette per PCR e puntali per pipette dovrebbero tutti essere obbligatoria bassa per minimizzare la perdita di DNA.
Nota: Condurre un'omogeneizzazione più presto, per la rapida essiccazione o morte dell'animale in modo significativo possa danneggiare il DNA genomico.
3. omogeneizzazione ed estrazione del DNA
- Omogeneizzare l'animale per ottenere il suo DNA genomico (gDNA) con uno dei seguenti metodi.
Nota: È fondamentale usare il kit specificato mostrato in Tabella materiali nelle seguenti operazioni di estrazione del DNA, per altri kit (colonna e perlina basata) non sono efficaci a questo ingresso estremamente basso. Il buffer di lisi genomico è stato fornito con 0,5% beta-mercaptoetanolo prima dell'uso.- Omogeneizzare l'animale con cicli di gelo e disgelo18.
- Subito dopo passo 2.5, aggiungere 100 µ l di tampone di lisi nella provetta di PCR contenente il Tardigrada.
- Collocare la provetta PCR in azoto liquido per 10 minuti e, per 10 min, spostarlo in un blocco di calore riscaldato a 37 ° C. Ripetere questo passaggio x 2.
Nota: Questo passaggio può essere ripetuto quando l'omogeneizzazione non è sufficiente o può essere eseguita dopo la pigiatura manuale.
- Manualmente e schiacciare l'animale.
- Subito dopo passo 2.5, sotto un microscopio stereoscopico, schiacciare l'individuo con la punta di P10 della pipetta premendo l'animale contro la parete del tubo PCR e immediatamente aggiungere 100 µ l di tampone di lisi.
Nota: È fondamentale osservare questa procedura sotto un microscopio stereoscopico, perchè il Tardigrada può facilmente scivolare via e frantumazione inefficiente si tradurrà in un errore per estrarre gDNA. Assicurarsi che la cuticola tardigrade è rotta in modo che il buffer di lisi possa infiltrare l'organismo.
- Subito dopo passo 2.5, sotto un microscopio stereoscopico, schiacciare l'individuo con la punta di P10 della pipetta premendo l'animale contro la parete del tubo PCR e immediatamente aggiungere 100 µ l di tampone di lisi.
- Omogeneizzare l'animale con cicli di gelo e disgelo18.
- Incubare la provetta per 30 min a temperatura ambiente per lisi si verifichi.
Nota: Un minimo di 30 minuti è necessario per un efficiente Lisi, ma l'incubazione può essere più lunga. - Trasferire il volume completo (100 µ l) della miscela Lisi ad una pulito 1,5 mL basso-associazione microprovetta.
- Aggiungere 100 µ l di tampone di lisi per il tubo PCR di basso-associazione che è stato utilizzato per l'omogeneizzazione e ora è vuoto e dopo la miscela di pipettaggio, trasferirlo nella microprovetta basso-associazione 1,5 mL utilizzato al punto 3.3. Ripetere questo passaggio x 2.
- Come descritto al punto 3.4, aggiungere 300 µ l di tampone di lisi per la provetta PCR bassa-associazione e, dopo il pipettaggio, spostare la miscela nella microprovetta 1,5 mL basso-associazione. I conetti di basso-associazione di 1,5 mL dovrebbero contenere 600 µ l della miscela dopo questo passaggio.
Nota: Questi passaggi sono per minimizzare la perdita di gDNA vincolato alla parete del tubo PCR, il campione di lavaggio più volte. - Aggiungere il totale di 600 µ l di miscela di Lisi a colonna spin inserito in un tubo di raccolta e centrifugare esso a 10.000 x g per 1 min.
- Applicare nuovamente il flusso attraverso la colonna ed e centrifugare a 10.000 x g per 1 min.
Nota: Questo passaggio è fondamentale per garantire che la maggior parte del gDNA è associato alla colonna. - Aggiungere 500 µ l di tampone di lavaggio per la colonna di spin e centrifugare a 10.000 x g per 1 min. trasferimento della colonna di spin ad una microprovetta 1,5 mL pulito.
- Applicare 20 µ l di 10 mM Tris-HCl, pH 8.5, alla colonna di spin, attendere 5 min a temperatura ambiente e centrifugare esso a 10.000 x g per 1 min.
Nota: Il tampone di eluizione non deve contenere EDTA, perché interferisce con gli enzimi di preparazione di biblioteca. - Applicare nuovamente il flusso attraverso la colonna di spin e dopo 5 min di incubazione a temperatura ambiente, e centrifugare per 1 min a 10.000 x g.
Nota: Questo passaggio è fondamentale per garantire la massima eluizione di gDNA associato alla colonna.
4. sequenza di costruzione biblioteca
-
Frammentazione del DNA
- Trasferire 15 µ l di eluente il gDNA una microprovetta 15-µ l per frammentazione del DNA e centrifugare la provetta per 1 min utilizzando una microcentrifuga da tavolo.
Nota: Nella microprovetta specificata mostrata in Tabella materiali è ottima per un basso input. La frammentazione con basso volume sonicazione è fondamentale, e questo non può essere sostituito con frammentazione enzimatica a causa della bassissima concentrazione di DNA. - Frammentare il gDNA a 550 bp.
Nota: Le impostazioni di bp 550 abbiamo usato sono come segue: potenza incidente picco = 30 W, fattore di utilizzo = 20%, cicli al burst = 50, tempo di trattamento = 23 s. - Dopo un'approfondita di pipettaggio, trasferire 10 µ l di miscela di DNA frammentata in una provetta PCR di pulito basso-associazione.
Nota: L'esperimento può essere fermato qui. Conservare il DNA a 4 ° C o -20 ° C.
- Trasferire 15 µ l di eluente il gDNA una microprovetta 15-µ l per frammentazione del DNA e centrifugare la provetta per 1 min utilizzando una microcentrifuga da tavolo.
-
Sequenza di costruzione di libreria
Nota: È assolutamente fondamentale per utilizzare il kit specificato nella Tabella materiali nelle procedure seguenti, a causa della quantità bassa di ingresso del DNA.- Aggiungere 2 µ l di tampone di preparazione del modello e 1 µ l di enzima di preparazione del modello e mescolare accuratamente con una pipetta.
- Eseguire la reazione di preparazione del modello in un termociclatore con le seguenti condizioni: 22 ° C per 25 min, 55 ° C per 20 min, presa a 4 ° C e un coperchio riscaldato a 101-105 ° C. Una volta completata la reazione, procedere al passaggio successivo.
- Aggiungere 1 µ l di buffer sintesi libreria e 1 µ l di enzima di sintesi la libreria per il prodotto di reazione di preparazione di modello e incubare la miscela a 22 ° C per 40 min (con una tenuta di 4 ° C). Una volta completata la reazione, procedere al passaggio successivo.
- Aggiungere 30 µ l di mix master biblioteca amplificazione (25 µ l di tampone di amplificazione la biblioteca, 1 µ l di enzima amplificazione libreria e 4 µ l di acqua priva di nucleasi) e 5 µ l di reagente di indicizzazione.
- Mescolare il tutto accuratamente con una pipetta e centrifugare la miscela con una centrifuga da tavolo.
- Eseguire una PCR con le condizioni presentate nella tabella 1.
| Temperatura | Tempo | Cicli |
| 72 ˚ c | 3 minuti | |
| 85 ˚ c | 2 minuti | |
| 98 ° C | 2 minuti | |
| 98 ° C | 20 secondi | 4 cicli |
| 67 ˚ c | 20 secondi | |
| 72 ˚ c | 40 secondi | |
| 98 ° C | 20 secondi | 16 cicli |
| 72 ˚ c | 50 secondi | |
| 4 ˚ c | Tenere premuto |
Tabella 1: Condizioni di PCR.
-
Purificazione dei prodotti di PCR
Nota: Questo passaggio può essere sostituito con altri metodi di purificazione. Se viene utilizzato un metodo alternativo, assicuratevi di controllare per la purezza di DNA risultante utilizzando uno spettrofotometro. Rapporti di assorbanza 260/280 e 260/230 devono essere entrambi sopra 1.8.- Aggiungere 50 µ l di biglie magnetiche, dispensare la soluzione 10 x e centrifugare con una microcentrifuga da tavolo.
- Incubare la soluzione per 2 min a temperatura ambiente.
- Incubare su un supporto magnetico per 5 minuti o fino a quando la soluzione diventa completamente cancellare e rimuovere il surnatante.
- Aggiungere 200 µ l di etanolo di 80% preparati al momento nella provetta PCR di basso-associazione su un supporto magnetico, attendere per 30 s e rimuovere il surnatante. Ripetere questo passaggio x 2. Non disturbare le perline.
- Brevemente, centrifugare la provetta PCR bassa-associazione con una centrifuga da tavolo e rimuovere qualsiasi etanolo in eccesso sul supporto magnetico. Asciugare le perline, ma evitare di seccare.
- Risospendere le perline con 15 µ l di 10 mM Tris-HCl, pH 8.5, accuratamente Pipettare la soluzione in modo che i branelli magnetici sono distribuiti in modo omogeneo, incubare la provetta per 2 min a temperatura ambiente e dopo una breve centrifugazione, incubare su un supporto magnetico per 2 min fino a quando la soluzione è limpida.
Nota: Il tampone di eluizione non deve contenere EDTA, perché interferisce con la chimica di sequenziamento. - Trasferire il surnatante, senza disturbare il pellet, ad un nuovo tubo PCR di basso-associazione.
Nota: L'esperimento può essere fermato qui. Conservare il DNA a 4 ° C o a-20 ° C per lo stoccaggio di lunga data.
5. qualità di controllo, la quantificazione e la sequenza del DNA
Nota: Un controllo di qualità non viene eseguito prima che questo passo a causa della bassa quantità di DNA.
-
Convalida della distribuzione di dimensione di biblioteca del DNA
Nota: Altri sistemi di elettroforesi ad alta sensibilità con una segnalazione digitale di distribuzione delle dimensioni possono essere utilizzati.- Restituire il reagente tampone di elettroforesi e gel cartuccia (25-1.000 bp nella gamma) a temperatura ambiente.
- Aggiungere 3 µ l di reagente tampone di elettroforesi con 1 µ l della libreria di sequenziamento e mescolare accuratamente per 1 min con un vortice e li Centrifugare brevemente con una centrifuga da tavolo.
- Condurre l'elettroforesi e convalidare la distribuzione di dimensione di biblioteca con il software associato. Il picco di frammento principale dovrebbe essere largamente vanno da circa 300 a 1.000 bp.
-
Quantificazione del DNA
Nota: Altri metodi basati sulla fluorescenza o PCR quantitativa può anche essere utilizzato, ma spettrofotometria dovrebbe essere evitato, in quanto non è sufficientemente accurata per quantificare le librerie di sequenziamento.- Aggiungere 796 µ l di soluzione tampone e 4 µ l di reagente fluorescente e mescolare accuratamente. Erogare 190 µ l di soluzione di lavoro di due provette e 197 µ l in una provetta di analisi.
- Aggiungere 10 µ l di standard con concentrazioni note di DNA a ciascuna provetta di analisi contenente 190 µ l di soluzione di lavoro e 3 µ l della biblioteca preparata nella provetta di analisi contenente 197 µ l di soluzione di lavoro.
- Vortex le provette brevemente e centrifugare li su una centrifuga da tavolo. Quantificare il DNA utilizza un fluorometro con impostazioni 3-µ l.
-
Sequenza di DNA biblioteca
Nota: La piattaforma di sequenziamento deve essere compatibile con il kit di costruzione libreria specificata.- Preparare la libreria di sequenziamento basata sul protocollo del produttore.
- Impostare la cella di cassette e flusso di reagente nello strumento sequenziamento e immettere la sequenza di esecuzione informazioni seguendo il protocollo del produttore.
- Eseguire il sequenziamento.
Nota: Abbiamo condotto due esecuzioni di sequenziamento: un esempio di esecuzione, come pure quattro campioni multiplexati in una corsa.
6. analisi computazionale
- Chiamata di base e, se necessario, demultiplex le letture.
- Convalidare la qualità dei dati di sequenza con FastQC19.
Nota: Per una convalida più approfondita dei dati ottenuti, Vedi il nostro precedente rapporto16.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Esclusione delle sostanze contaminanti:
Questo protocollo prevede un lavaggio accurato del Tardigrada e una sterilizzazione con antibiotici cura per minimizzare la contaminazione. Implica anche un processo di verifica visivo per assicurare la completezza di questi processi. Un'immagine del microscopio fatta durante la convalida (punto 2.4 del protocollo) è illustrata nella Figura 2. Quando osservate un ingrandimento X 500, cellule batteriche possono essere visto come piccole particelle che si muovono intorno il Tardigrada individuo.
Convalida della qualità libreria del DNA:
L'importo totale della libreria del DNA-Seq costruita è circa 109.5 ng (7,3 ng / µ l x 15 µ l)16. Per convalidare la distribuzione di lunghezza della frammentazione, un modello di elettroforesi dovrebbe essere simile a nella figura 3. Come abbiamo impostato la dimensione di frammentazione a 550 bp con un DNA sistema di taglio, la biblioteca dovrebbe essere bp di 550-600, tra cui gli adattatori di sequenziamento. Si può osservare che la maggior parte della libreria di sequenza è contenuta tra 200-1000 bp ed è coerenza tra repliche (N1 - N4).
Analisi di dati di sequenza:
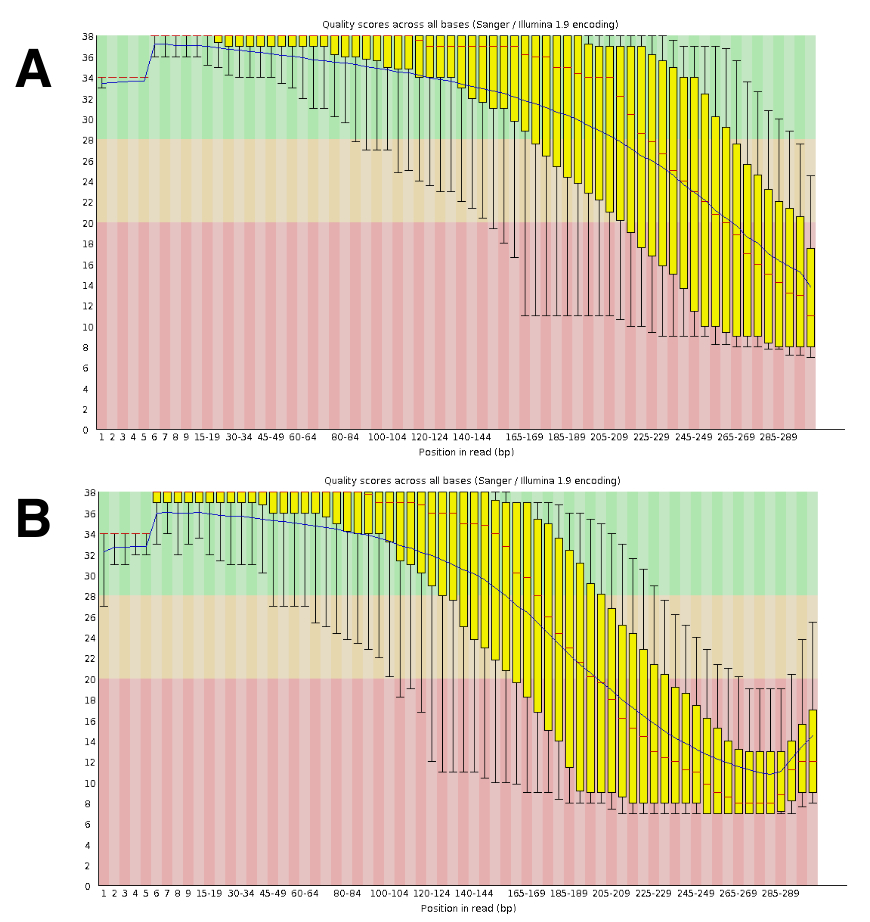
Il sequenziamento del DNA generato circa 20 a 25-M letture accoppiate per corsa. La convalida della qualità è stata condotta utilizzando FastQC (Figura 4). La distribuzione della qualità lungo la lettura in sequenza è tipica di una corsa di 300-bp accoppiata.

Figura 1: flusso di lavoro del presente protocollo. Questa figura mostra un riepilogo di questo protocollo. Clicca qui per visualizzare una versione più grande di questa figura.

Figura 2 : Foto rappresentativa di un Tardigrada esente da batteri. Questa figura mostra immagini di un contaminati Tardigrada (sinistra) e pulito (a destra) Tardigrada (Hypsibius dujardini), insieme a ulteriori immagini ingrandite (in basso). Asta-a forma di cellule intorno il Tardigrada sono contaminanti e sono indicate con una freccia. La barra della scala indica 100 µm. Clicca qui per visualizzare una versione più grande di questa figura.

Figura 3 : Convalida della distribuzione di lunghezza di frammento della biblioteca del DNA costruita. Questo pannello mostra la distribuzione della dimensione di biblioteca del sequenziamento. Le linee viola e verde indicano i marcatori superiori e inferiori a 1.500 e 25 bp, rispettivamente. L = Ladder, S = 1 campione/Esegui, N1 - N4 = 4 test/serie. Clicca qui per visualizzare una versione più grande di questa figura.

Figura 4 : Esempio della convalida della qualità del DNA-Seq con FastQC. A FastQC per convalidare le prestazioni di sequenza sono stati presentati dati DNA-Seq. Un risultato rappresentativo per DRR055040 per qualità di sequenze di base è mostrato (DDBJ sequenza lettura archivio DRA00445516). (A), questo pannello mostra le letture in avanti (R1). (B) questo pannello mostra le letture inversione (R2). Clicca qui per visualizzare una versione più grande di questa figura.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Contaminazione batterica rappresenta una minaccia per il sequenziamento genomico di organismi microscopici. Mentre gli studi precedenti sul sequenziamento del genoma tardigrade hanno filtrato contaminazione utilizzando metodi informatica vasto12,20, abbiamo sequenziato il genoma da un singolo individuo per ridurre al minimo il rischio di contaminazioni. Poiché un singolo Tardigrada contiene circa il 50-200 pg di genomic DNA16 ed è racchiuso in uno spesso strato di esoscheletro di chitina, l'esclusione delle sostanze contaminanti ed estrazione di DNA di alta qualità sono i punti critici in questo protocollo. Non esistenti i tardigradi culture sono asettiche e quelli raccolti dal selvaggio trasporta un sacco di contaminanti sulla superficie, come pure i resti di cibo nei loro intestini. Precedenti progetti di sequenziamento del genoma di tardigradi hanno sequenziato 10.000-100.000 individui collettivamente come un campione12,14, che significa che i risultati sono molto suscettibili di essere influenzati da contaminanti batterici. Nella loro relazione, Boothby et al. raccolti individui H. dujardini utilizzando il loro comportamento di fototassi negativo14, e il gruppo non impiegava metodi anti-batterico.
Per esaminare visivamente se ci sono contaminanti, abbiamo incubato Tardigrada in antibiotici (penicillina/streptomicina) ed abbiamo esaminato l'individuo sotto un microscopio X 500. Isolando un singolo individuo e ispezionarlo accuratamente per eventuali contaminanti, abbiamo minimizzato il rischio di possibili contaminazioni. Bassi livelli di contaminazione sono stati confermati dai dati di sequenziamento come ben16. Per quanto riguarda l'estrazione del DNA, abbiamo impiegato omogeneizzazione manuale, come pure di omogeneizzazione termica18. Inviando all'individuo tardigrade di azoto liquido e 37 ° C, le crepe sono state indotte nell'esoscheletro di chitina e il buffer di lisi era in grado di penetrare nel corpo e lisare le cellule. Quando il rendimento del DNA rimane più basso del previsto sia manuale che termico omogeneizzazione può essere condotta per massimizzare il rendimento.
Il metodo indicato in questo articolo presenta diverse limitazioni. In primo luogo, omogeneizzazione di cicli di gelo e disgelo è stato applicato da uno studio sui nematodi; quindi, il metodo può solo essere efficace contro ecdysozoa. In secondo luogo, per l'amplificazione dei frammenti del DNA durante la fase di raccolta di sequenza del DNA, la possibilità di errori PCR non può essere ignorata. Pertanto, i dati di sequenza non sono raccomandati per l'analisi che richiede alta precisione letture (cioè, analisi SNP). Inoltre, come abbiamo dichiarato nel protocollo, l'utilizzo del kit SNA-Seq specificato mostrato in Tabella materiali è assolutamente critica, a causa della quantità bassa di ingresso del DNA. Questo kit di costruzione libreria DNA lega sequenze adattatore Illumina prima l'amplificazione; Pertanto, questa libreria non può essere applicata per il sequenziamento di lungo-lettura utilizzando la tecnologia PacBio o Nanopore. Infine, un controllo di qualità della libreria DNA costruito durante questo protocollo si verifica una sola volta, dopo la costruzione della libreria di sequenziamento. Ciò è dovuto il basso input del DNA dal maggior parte quantificazione del DNA e metodi di elettroforesi non possono rilevare il 50-200 pg di DNA. Di conseguenza, abbiamo condotto controlli di qualità, quali l'elettroforesi (Figura 1) e la fluorescenza-basata quantificazioni, solo dopo l'amplificazione di PCR.
Una trattazione completa delle analisi bioinformatica dei dati esula dall'ambito del presente articolo; Tuttavia, abbiamo brevemente detto parecchie analisi che abbiamo condotto. Un controllo di qualità dei dati di sequenziamento con FastQC19 calcola le qualità per-base, duplicazione di sequenza, i dati di sequenza ecc che sono stati convalidati possono essere presentate all'Assemblea del genoma. Ci hanno montato un genoma di 132 Mb con MaSuRCA v 3.1.321 e hanno confrontato le statistiche di mappatura calcolate con BWA22 e QualiMap23 di questa libreria di sequenziamento del DNA con altri H. dujardini genoma assembly16. Inoltre, abbiamo anche usato questo dati di sequenziamento del DNA per l'esclusione di contaminanti nel nostro Studio17e hanno osservato che le letture in sequenza sono distribuite uniformemente in tutto il genoma.
Maggior parte dei progetti su organismi non-modello a partire da coltura abbastanza materiale di esempio, come è avvenuto con i tardigradi24. Progressi tecnici in tecniche di coltura hanno permesso di elevate quantità di tardigrade cultura, ma metodi di coltura attuale non sono ancora asettici e poiché la maggior parte i tardigradi sono ancora VBNC nei laboratori, è stato quasi impossibile per condurre il genoma o sequenziamento del trascrittoma. Questo metodo di sequenziamento del DNA da un singolo individuo rende possibile l'analisi di specie tardigrade rare, tra cui specie marine che sono stati studiati in meno. Conducendo genomica comparata a una più ampia area gradualismo, può essere realizzata un'ulteriore comprensione dei meccanismi anhydrobiosis nei tardigradi.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Gli autori non hanno nulla a rivelare.
Acknowledgments
Gli autori ringraziano Nozomi Abe, Yuki Takai e Nahoko Ishii per il loro supporto tecnico nel sequenziamento genomico. Quest'opera è stata sostenuta di sovvenzione per la società del Giappone per la promozione della scienza (JSPS) Research Fellow, sovvenzione dai KAKENHI per giovani scienziati (No.22681029) e KAKENHI sovvenzione per la ricerca scientifica (B), no. 17 H 03620 da JSP, da un Sovvenzione per progetti di ricerca di scienza base da The Sumitomo Foundation (No.140340) e in parte da fondi di ricerca dal governo della Prefettura di Yamagata e città di Tsuruoka, Giappone. Chlorella vulgaris utilizzata per alimentare i tardigradi è stato fornito per gentile concessione di clorella Industry Co. Ltd.
Materials
| Name | Company | Catalog Number | Comments |
| SZ61 microscope | OLYMPUS | ||
| BactoAgar | Difco Laboratories | 214010 | |
| Penicillin Streptomycin (10,000 U/mL) | Gibco by life technologies | 15140-148 | |
| VHX-5000 System | Keyence | ||
| 0.2mL Silicone coating tube | Bio Medical Science | BC-bmb20200 | |
| Quick-DNA Microprep Kit | ZYMO Research | D3021 | Use of this kit is absolutey critical; see step 3.1 |
| 1.5 mL microtube | greiner bio-one | 616-201 | See 4.1.1 |
| HIgh speed refrigerated micro centrifuge | TOMY | MX-307 | |
| Covaris M220 | Covaris Inc. | 4482277 | |
| ThruPLEX DNA-Seq kit | Rubicon Genomics | CAT. NO. R400406 | Use of this kit is absolutey critical; see step 4.2 |
| Thermal Cycler | Bioer Technology | TC-96GHbC | |
| AMPure XP reagent | BECKMAN COULTER Life Science | A63881 | |
| Ethanol | Wako | 054-027335 | |
| EB buffer | QIAGEN | 19086 | |
| 2200 TapeStation | Agilent | G2965AA | |
| D1000 Reagents | Agilent | 5067-5583 | |
| D1000 ScreenTape | Agilent | 5067-5582 | |
| Qubit dsDNA BR Buffer/Reagent | ThermoFisher Scientific | Q32850 | |
| Cubee Mini-Centrifuge | RecenttecGenereach | R5-AQBD01aqbd | |
| MiSeq 600 cycle v3 | Illumina Inc. | MS-102-3003 | |
| MiSeq Sequencer | Illumina Inc. | SY-410-1003 |
References
- Crowe, J. H., Hoekstra, F. A., Crowe, L. M. Anhydrobiosis. Annual Review of Physiology. 54 (1), 579-599 (1992).
- Mobjerg, N., et al. Survival in extreme environments - on the current knowledge of adaptations in tardigrades. Acta Physiologica. 202 (3), 409-420 (2011).
- Becquerel, P. La suspension de la vieau dessous de 1/20 K absolu par demagnetization adiabatique de L'alun de fer dans le vide les plus eléve. Comptes Rendus de l'Académie des Sciences. 231, 261-264 (1950).
- Ono, F., et al. Effect of ultra-high pressure on small animals, tardigrades and Artemia. Cogent Physics. 3 (1), 1167575 (2016).
- Horikawa, D. D., et al. Tolerance of anhydrobiotic eggs of the Tardigrade Ramazzottius varieornatus to extreme environments. Astrobiology. 12 (4), 283-289 (2012).
- Horikawa, D. D., et al. Analysis of DNA repair and protection in the Tardigrade Ramazzottius varieornatus and Hypsibius dujardini after exposure to UVC radiation. PLoS One. 8 (6), e64793 (2013).
- Horikawa, D. D., et al. Radiation tolerance in the tardigrade Milnesium tardigradum. International Journal of Radiation Biology. 82 (12), 843-848 (2006).
- May, R. M., Maria, M., Gumard, J. Action différentielle des rayons x et ultraviolets sur le tardigrade Macrobiotus areolatus, a L'état actif et desséché. Bulletin Biologique de la France et de la Belgique. 98, 349-367 (1964).
- Jonsson, K. I., Harms-Ringdahl, M., Torudd, J. Radiation tolerance in the eutardigrade Richtersius coronifer. International Journal of Radiation Biology. 81 (9), 649-656 (2005).
- Bemm, F., Weiss, C. L., Schultz, J., Forster, F. Genome of a tardigrade: Horizontal gene transfer or bacterial contamination? Proceedings of the National Academy of Sciences of the United States of America. 113 (22), E3054-E3056 (2016).
- Delmont, T. O., Eren, A. M. Identifying contamination with advanced visualization and analysis practices: metagenomic approaches for eukaryotic genome assemblies. PeerJ. 4, e1839 (2016).
- Koutsovoulos, G., et al. No evidence for extensive horizontal gene transfer in the genome of the tardigrade Hypsibius dujardini. Proceedings of the National Academy of Sciences of the United States of America. 113 (18), 5053-5058 (2016).
- Boothby, T. C., Goldstein, B., et al. Reply to Bemm et al. and Arakawa: Identifying foreign genes in independent Hypsibius dujardini genome assemblies. Proceedings of the National Academy of Sciences of the United States of America. 113 (22), E3058-E3061 (2016).
- Boothby, T. C., et al. Evidence for extensive horizontal gene transfer from the draft genome of a tardigrade. Proceedings of the National Academy of Sciences of the United States of America. 112 (52), 15976-15981 (2015).
- Arakawa, K. No evidence for extensive horizontal gene transfer from the draft genome of a tardigrade. Proceedings of the National Academy of Sciences of the United States of America. 113 (22), E3057 (2016).
- Arakawa, K., Yoshida, Y., Tomita, M. Genome sequencing of a single tardigrade Hypsibius dujardini individual. Scientific Data. 3, 160063 (2016).
- Yoshida, Y., et al. Comparative genomics of the tardigrades Hypsibius dujardini and Ramazzottius varieornatus. PLoS Biology. 15 (7), e2002266 (2017).
- He, F. Total RNA Extraction from C. elegans. Bio-protocol. Bio101, e47 (2011).
- Andrews, S. FastQC a quality-control tool for high-throughput sequence data. , http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (2015).
- Hashimoto, T., et al. Extremotolerant tardigrade genome and improved radiotolerance of human cultured cells by tardigrade-unique protein. Nature Communications. 7, 12808 (2016).
- Zimin, A. V., et al. The MaSuRCA genome assembler. Bioinformatics. 29 (21), 2669-2677 (2013).
- Li, H., Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 25 (14), 1754-1760 (2009).
- Okonechnikov, K., Conesa, A., Garcia-Alcalde, F. Qualimap 2: advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics. 32 (2), 292-294 (2016).
- Horikawa, D. D., et al. Establishment of a rearing system of the extremotolerant tardigrade Ramazzottius varieornatus: a new model animal for astrobiology. Astrobiology. 8 (3), 549-556 (2008).

{kind=link}