1. Cell Seeding
NOTE: The described cell spreading protocol was performed using mouse embryonic fibroblasts (MEFs) expressing PH-Akt-GFP (a fluorescent marker for PIP3/PI(3,4)P2). This cell line was generated by genomically integrating an expression construct for PH-Akt-GFP (Addgene #21218) by CRISPR-mediated gene editing. However, other fluorescent markers that are expressed transiently or integrated in the genome can also be used in this assay. For optimal image segmentation, we recommend using fluorescent markers that are evenly distributed in the cytoplasm, e.g., cytosolic GFP.
- Culture a 10 cm dish of cells to 90% confluency.
- Once the cells have attained the proper confluency, place a 22 mm x 22 mm coverslip (#1.5; 0.17 mm thickness) into a 35 mm cell culture dish. Coat the coverslip with 400 µL of fibronectin that has been diluted in PBS to a final concentration of 2.5 µg/mL.
NOTE: The number of coverslips required for the assay is determined by the number of experimental conditions and technical replica. - Place the 35 mm dish with the fibronectin-coated coverslip into a 37 °C, 5% CO2 incubator for 1 hour.
- Remove the dish with the coverslip from the incubator. Aspirate the fibronectin and wash the coverslip with PBS by gently pipetting around the coverslip two to three times.
- Aspirate the cell culture media from the 10 cm dish of cells and wash the dish with PBS.
- Add 650 µL of 0.05% trypsin-EDTA to the 90% confluent dish of cells, tilting the dish to evenly distribute the enzyme. Place the dish with the trypsin into the incubator for 1 minute.
- Remove the dish with the cells from the incubator. Add 10 mL of cell culture media into a 15 mL centrifuge tube. Quickly add another 10 mL of media into the dish to quench the trypsin.
- Pipette 1 mL of the trypsinized cells into the 15 mL centrifuge tube in order to dilute the cells. Pipette the contents of the tube up and down to ensure an even distribution of cells within the media. For cell types with high aggregation propensity, filtering cells through a cell strainer (100 µm mesh size) is recommended to minimize the occurrence of cell clumping.
- From the tube, pipette 500 – 1000 µL of diluted cells into the 35 mm dish containing the coverslip.
- Gently shake the dish to evenly spread out the cells. Ensure that the coverslip is at a ~10% confluency (~50,000 cells/mL) and adjust the volume of diluted cells as needed.
NOTE: The purpose of having cells at such low confluency is to ensure that there are 1-2 polarized cells in each field of view that will be used to focus the objective throughout the cell spreading acquisition. - Passage 1/5 of the remaining cells in the 10 cm dish into one 6 cm dish per treatment condition. Place the passaged dishes and the 35 mm dish with the coverslip into the incubator overnight.
NOTE: These will be the cells that will be analyzed for spreading dynamics.
2. Drug Incubation and Cell Recovery
- Add 5 mL of cell culture media into each of two 15 mL centrifuge tubes, and 20 mL of phenol red free DMEM into each of two 50 mL centrifuge tubes.
NOTE: The number of tube pairs (15 mL + 50 mL) should correspond to the number of experimental conditions. - To test the importance of Arp2/3 for cell spreading, pipette either the pharmacological inhibitor of Arp2/3, CK-666, or the control treatment, such as DMSO, into each pair of centrifuge tubes up to the desired concentration.
- Remove the passaged 6 cm dishes (see Step 1.11) from the incubator and aspirate the media. Wash the dishes with warm PBS.
- Add the contents of the CK-666- or DMSO-supplemented 15 mL centrifuge tubes into each of the passaged dishes. Label each dish with the correct drug treatment and place the dishes into the incubator for one hour.
- Remove the dishes from the incubator and aspirate the media. Wash the dishes with warm PBS in order to thoroughly remove all the remaining phenol red media.
- Add 230 µL of 0.05% trypsin-EDTA to each 6 cm dish and incubate cells for 1 minute.
NOTE: If applicable, trypsin can be replaced by a non-proteolytic cell adhesion blocker. - Remove the dishes from the incubator. For each treatment, add 5 mL of drug-supplemented phenol red free DMEM into a 15 mL centrifuge tube designated as "Tube B". Add an additional 5 mL of the same media into the relevant dish to quench the trypsin. Transfer the contents of the dish into a 15 mL centrifuge tube designated as "Tube A".
- Transfer 1 mL of cells from Tube A into Tube B. Repeat for each treatment.
- Place Tubes A and B into the incubator for 45 minutes to allow cells to recover from trypsinization. Slightly loosen the cap of the centrifuge tubes before placing them in the incubator to allow for CO2 penetrance.
NOTE: The duration of recovery time may vary for different cell types. Although in our experiments 45-minute-long recovery had a negligible effect on cell viability, some cell types may undergo anoikis when maintained in suspension for too long. Therefore, we recommend determining the optimal recovery time empirically. The optimal recovery time enables fast and synchronous cell spreading with no dead or apoptotic cells in the sample.
3. Magnetic Chamber Preparation
- Ensure that all parts of a 1 Well Chamlide Cell Magnetic Chamber that can accommodate a 22 mm x 22 mm square coverslip have been cleaned before use.
- Remove the 35 mm dish with the coverslip (see Step 1.11) from the incubator. Aspirate the cell culture media and wash the coverslip with warm PBS.
- Remove the coverslip from the 35 mm dish using a pair of forceps and gently lay the coverslip onto the bottom plate of the magnetic chamber.
- Place the silicone gasket on top of the coverslip.
NOTE: An improperly placed silicone gasket is the most common cause of a leaky magnetic chamber. Ensure that the gasket rests in the indent of the bottom plate and does not rise beyond the indent. - Attach the main body onto the bottom plate.
NOTE: Do this part very slowly. A good tip is to hold down the bottom plate with one hand while placing the main body on top. This ensures that the main body's magnets do not lift the bottom plate up, which could potentially displace and crack the coverslip. - Add 1 mL of drug-supplemented phenol red free DMEM to the magnetic chamber. Take a lint-free tissue and carefully dab the enclosure between the main body and the bottom plate in order to check for any leaks.
NOTE: If there is leakage, quickly aspirate the media and proceed again from step 3.4. - Lower the transparent cover onto the main body to enclose the magnetic chamber.
- Spray a laboratory tissue first with water and wipe the bottom of the magnetic chamber (the coverslip, not the metal part). Afterwards, spray a second laboratory tissue with a small amount of 70% ethanol and wipe, being careful not to crack the coverslip.
4. Image Acquisition
- Preheat the stage top incubator and the objective heater to 37 °C and set the CO2 level in the stage top incubator to 5%.
NOTE: If the stage top incubator is not connected to a CO2 supply, the cell culture media should be supplemented with 25 mM HEPES to maintain constant pH 7.4. - Apply a sufficient amount of immersion oil to the pre-warmed 60X, 1.4 N.A. oil objective.
NOTE: We use a 60X, 1.4 N.A. oil immersion objective in this protocol because of its reasonably large field of view and outstanding light collection efficiency. If a larger field of view is required, a lower magnification objective (e.g., 20x) can be used as long as the signal-to-noise ratio of the images is greater than 2.5. - Bring both the completed magnetic chamber and Tube B (Step 2.9) to the confocal microscope. Place the magnetic chamber onto the stage top incubator.
NOTE: Place the magnetic chamber gently on the stage to avoid creating bubbles in the immersion oil. - Set focus to the fluorescent cells using the GFP channel. Make sure that the cell edge is sharp and well defined.
- Remove the transparent cover of the magnetic chamber and pipette 500 µL from Tube B into the magnetic chamber. Place the transparent cover back on top of the magnetic chamber.
- To identify cells ideal for cell spreading analysis, search for "halos" of cells that have yet to attach to the coverslip but are no longer rolling around. Cells that are in the earliest stages of coverslip attachment are also great candidates, but image acquisition must be swift in order to capture spreading.
- Configure the time-lapse image acquisition for the green channel to include four fields of view, imaged at 6 second intervals.
NOTE: Due to the high variability of lamellipodia protrusion velocity among different cell types, the optimal frame rate should be determined empirically. The imaging interval of 6 seconds used in our experiments is a good starting point for the analysis of many mesenchymal and epithelial cells. However, cells that spread very quickly (e.g., immune cells) may require a much higher frame rate (shorter imaging interval). The optimal frame rate for cell spreading movies ensures a 2-5 pixels displacement of the protruding cell edge between subsequent frames. Considering the accuracy of curve fitting used to identify the plateau of cell spreading, the optimal frame rate should also ensure 50-100 measurements of cell edge displacement during the rapid expansion phase of cell spreading. The number of fields of view should be adjusted depending on the exposure time, the distance between acquisition points, and the stage movement speed. Users are advised to determine the maximum number of fields of view that can acquired with the desired frame rate. - After identifying a suitable field of view, save the X and Y coordinates of the microscope stage. Proceed with identifying three other fields of view that are relatively close to one another on the coverslip. Save the coordinates of the microscope stage for every desired field of view.
NOTE: It is strongly recommended to optimize the stage movement path between fields of view in order to minimize any unnecessary sample movement. Such optimization can be performed either manually or automatically. Excessive sample movement slows down the acquisition and may cause cells to roll out of view as they are descending. - Acquire images for 15 minutes at a 6 second frame rate and save the files. If more acquisitions are required, repeat starting at Step 4.6.
5. Analysis of cell area, circularity and protrusion dynamics during cell spreading
- Prepare images for data processing and analysis
NOTE: The software requires an image in .tiff format and a pixel size as the input parameters. Both requirements can be fulfilled using the acquisition software or Fiji (in this protocol). If these requirements are fulfilled, proceed to step 5.2.- Install the latest version of Fiji application (https://imagej.net/Fiji/Downloads).
- Open a time-lapse image using Fiji.
- Copy the pixel size of the image by selecting Image > Properties. Copy and paste the pixel size in µm to Notepad/Word.
- For the analysis of cell spread area and circularity, save the time-lapse image as a tiff image stack. The custom-build analysis software does not support proprietary file formats. Save the individual cell tiff image stack by selecting File > Save As > Tiff.
- Install the Python IDE (Spyder) and the necessary packages (PySimpleGUI and tifffile) for data processing and analysis.
NOTE: The installation of Python and packages is only required for the initial setup.- The time-lapse movies will be analyzed in the Spyder IDE using a custom-build Python script. To download the Spyder IDE, download the Anaconda distributor (https://www.anaconda.com/products/individual) which includes Spyder IDE and most of the necessary libraries and packages for this analysis.
- Install Anaconda and launch Spyder through Anaconda Navigator.
- In the IPython console tab (located in the lower right section of Spyder), copy and paste the following command: pip install PySimpleGUI and press the enter key. Running this command will install the package needed to initiate the graphical user interface (GUI).
- In the same console, copy and paste the following command: pip install tifffile and press the Enter key. Running this command will install the package needed to save images as tiff files.
- Download all the Python scripts from the supplemental files or the most updated scripts from GitHub at: https://github.com/ernestiu/Cell-spreading-analysis.git
- Quantify cell area and cell shape factors during cell spreading
- Open the main analysis script "cell_spreading_GUI.py" by selecting the open file option in the top panel of Spyder or using the shortcut Ctrl + O.
- Open the cell spreading analysis GUI by selecting "Run file" in the top panel or using the shortcut F5.
- Click the Cell Spread Area tab (Figure 3A).
- Select the tiff image to be analyzed using the browse button.
NOTE: The selected file must be a tiff file. - Specify the destination directory where data outputs (e.g., cell masks, values) will be saved.
- Specify the data output settings:
- Save masks: Save cell masks generated during the segmentation process.
- Export data: Export an excel spreadsheet (.xlsx) that contains all the analysis data to the destination folder.
Cell area, circularity and aspect ratio of all spreading cells will be saved as an Excel spreadsheet in the destination folder. The cell area is calculated as:
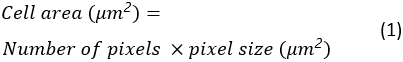
The cell circularity is a measure of how close a cell is to a perfectly round cell. It is calculated as:
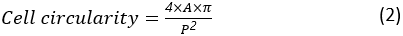
where A and P are the cell area and the cell perimeter, respectively. The aspect ratio of the cell represents how elongated the cell is. A spreading cell should have an aspect ratio close to 1. The aspect ratio is calculated as:
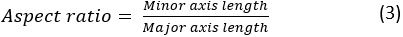
- Save contours: Save the cell boundary contour overlay images in the destination folder.
- Specify the segmentation settings
- Show segmentation: Show the segmentation result in the Spyder console during the analysis process.
- Smallest cell area (µm2): Enter the minimum value for cell area, including area values of cells in the beginning stages of attachment. Objects with an area smaller than this threshold will not be considered as spreading cells. This number will affect the segmentation process.
- Specify the image parameters.
- Acquisition interval (s): Enter the frequency of the image acquisition in seconds.
- Pixel size (µm): Enter the pixel size that was recorded when preparing images for analysis.
- Image bit depth: Enter the bit depth of the camera/detector.
- Click Run. If an error arises, an error message will appear in Spyder's console. Otherwise, the image analysis process will be shown in the console.
NOTE: The first image to appear in the console/Plots section (depending on the Spyder settings) shows all the cells identified in the field of view. Green boxes placed around the cells indicate spreading cells that are suitable for segmentation and analysis. Grey boxes indicate cells that are not suitable for analysis. The total number of identified spreading cells will also appear in the Console tab. The software plots the cell area (in blue) and cell circularity (in red) as a function of time. These graphs allow users to evaluate the accuracy of cell segmentation. A successful segmentation yields a monotonically increasing curve for cell area. To obtain a representative curve of cell spread area, the lag phase should be removed from the graph manually. The lag phase includes measurements of cell area before the cell starts spreading. The lag phase is indicated by fast fluctuations, as represented in the cell area plot (Figure 3C right).
6. Quantify cell edge dynamics during cell spreading using kymographs
- Prior to running the analysis, crop the raw movies of spreading cells to create time series of individual spreading cells.
- Use the Rectangle tool in the Fiji tool bar to manually select a region of interest (ROI) that encapsulates a single cell. (To ensure that the ROI fully encapsulates the spreading cell, use the scroll function to inspect the ROI at all time points.)
- Right click on the ROI and select Duplicate.
- Check Duplicate stack and click OK.
- Open the main analysis script "cell_spreading_GUI.py" by selecting the Open File button in Spyder's tool bar or using the shortcut Ctrl + O. If the GUI has already been opened, go directly to Step 6.3.
- Open the cell spreading analysis GUI by selecting Run file in the top panel or using the shortcut F5 (Figure 3B).
- Click the Kymograph generator & analysis tab.
- Use the browse button to select the tiff image for the analysis.
NOTE: Proprietary file formats, e.g., nd2, lif, zen, are not supported by the script. - Specify the destination folder to save the output data (cell masks and values).
- Specify the output settings.
- Export data: Export an excel spreadsheet (.xlsx) to the destination folder that contains relative cell edge positions and retraction events of the kymographs.
- Specify the image parameters:
- Acquisition interval (s): Enter the image acquisition frequency in seconds.
- Pixel size (µm): Enter the pixel size that was recorded when preparing images for the analysis in Step 5.1.3.
- Smallest cell area (µm2): Enter the minimum value for cell area, including area values of cells in the beginning stages of attachment. Objects with an area smaller than this threshold will not be considered as spreading cells. This number will affect the segmentation process.
- Image bit depth: Enter the bit depth of the camera/detector.
- Click Run. If an error arises, an error message will appear in Spyder's console. Otherwise, a summary of the protrusion dynamics quantifications will be shown in the console. There will be 4 pairs of retraction frequency and protrusion speed measurements, which are extracted from 4 kymographs generated from the top, bottom, left and right portions of the cell. The retraction frequency is calculated as:
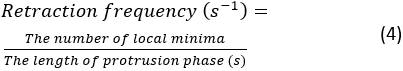
NOTE: This number demonstrates how frequently the lamellipodium retracts over the course of spreading. The average protrusion speed is measured by the slope between the beginning of protrusion and the plateau point on the kymograph. A summary kymograph figure will be shown in the console after the segmentation. To save the summary figure, right click on the figure and save the image.
The above protocol describes the experimental procedures for the live-cell imaging of spreading cells and a computational tool for the quantitative analysis of cell spreading dynamics. The computational tool can be used in a low- or high-throughput format to identify the molecular players regulating the actin polymerization machinery at the cell leading edge.
The schematic representation of the experimental procedures is depicted in Figure 1. The cell spreading assay was performed on immortalized mouse embryo fibroblasts stably expressing the pleckstrin homology (PH) domain of the Akt protein kinase tagged with eGFP25. The cells were detached with trypsin-EDTA and were allowed to recover in suspension for 45 minutes. During the recovery step, cells replenished their integrin receptors on the plasma membrane as indicated by the fast and synchronous attachment of the recovered cells to the fibronectin coated coverslips (Figure 2). Without the recovery period, spreading cells exhibited a broad distribution of cell size indicating a high variability in the onset of cell spreading (Figure 2A and B). Next, cells were plated on a fiducially-marked coverslip and their spreading dynamics were visualized by spinning disk confocal microscopy (schematics shown in Figure 1A – H). Throughout the image acquisition, we considered fields of view that featured cells with a signal-to-noise ratio of 2.5 or above. This was an important consideration as the subsequent image segmentation is sensitive to the cells' fluorescence intensity relative to the background. In our experiments, we acquired images every 6 seconds for 15 minutes (schematics shown in Figure 1I-J). In agreement with previous reports16, imaging at a 6 second frame rate ensured sufficient temporal resolution for capturing the dynamics of individual protrusion and retraction events, while allowing us to acquire several fields of view in parallel. The resulting time-lapse images were analyzed using the custom-build Python software (Figure 3).
An unbiased quantification of cell spreading was performed by using two distinct analytical procedures: (i) Morphodynamic profiling of spreading cells (Figure 3A, C and 4) and (ii) Kymograph analysis of cell edge dynamics (Figure 3B and 5). The analysis of cell spreading by morphodynamic profiling involves the automated detection of the spreading and fiducial cells in the field of view (Figure 3C left), followed by a frame-by-frame image segmentation and detection of the spreading cell boundary. The segmentation is performed by global intensity thresholding the individual frames. The threshold value is calculated as the local minimum between the first and second intensity modes on the image histogram26. Images with a unimodal, right-skewed histogram are segmented by the triangle thresholding algorithm27. Following cell segmentation, the morphodynamic characteristics of the spreading cells (i.e., cell area, aspect ratio, and cell circularity) were computed (Figure 3C right).
Consistent with published results12,28, cell spreading was driven by an isotropic expansion of lamellipodia as indicated by a sigmoidal shape on the representative cell area plot (Figure 3C right, blue curve and S1). The area plot showed that the cell area increased by approximately 3-fold before reaching a plateau (Figure 4A and B). Throughout the process of cell spreading, the cells remained circular (circularity = 0.70 ± 0.076) and displayed veil-like protruding cell edges, which are indicative of lamellipodial protrusions (Figure 4C).
To validate the cell spreading assay, we inhibited Arp2/3 with 100 µM CK-666 for 1 hour and 45 minutes and assessed the effect of this treatment on the cell spreading dynamics. In agreement with previous reports13, the suppression of Arp2/3 activity did not result in a significant decrease in the cell spreading speed (Figure 4A and B, pink curve). However, cell shape analysis revealed a significant difference in the circularity of control and CK-666 treated cells (Control: 0.70 ± 0.08 vs. CK-666: 0.54 ± 0.09, p < 0. 1 x 10-3) (Figure 4C). While the control cells remained circular until the plateau, Arp2/3-inhibited cells acquired a polygonal shape which was retained throughout the course of spreading (Figure 4A). Together, these experimental results demonstrate that the described cell spreading assay reveals moderate changes in cell morphodynamics caused by perturbations of the actin polymerization machinery.
While morphodynamic profiling is often sufficient to detect gross alterations in cell spreading dynamics, this analysis has limited ability to identify specific cytoskeletal components that regulate the protrusion-retraction cycles of the cell edge, prompting us to implement an unbiased Kymograph Analysis tool (Figure 5 left, dashed lines). The analysis of cell edge speed revealed a moderate but significant decrease in the average protrusion speed of Arp2/3-inhibited cells compared to control (Control: 37.1 ± 12.87 nm/s vs. CK-666: 28.7 ± 13.4 nm/s, p = 0. 9 x 10-3) (Figure 5C). Furthermore, dynamics of the cell edge of control and Arp2/3-inhibited cells were remarkably different (Figure 5A and D). The control cells protruded persistently with little to no retractions during the rapid expansion phase, which lasted about 200 seconds, and exhibited intermittent retractions during the plateau phase (Figure 5A and D). In contrast, the expansion of Arp2/3-inhibited cells was intervened by retraction events, denoted by the red dots on the graphs (Figure 5B and D). Quantification of retraction frequency showed that Arp2/3-inhibited cells retracted 26% more frequently than control cells (Control: 0.18 ± 0.22 s-1 vs. CK-666: 0.24 ± 0.19 s-1, p = 0.03) (Figure 5D). These data demonstrate the high sensitivity of the kymograph analysis in detecting mild alterations in lamellipodial dynamics.

Figure 1: The experimental workflow of a cell spreading assay. (A – H) Schematics of the cell spreading assay. (A) A 22 mm x 22 mm coverslip is coated with fibronectin diluted in PBS to a final concentration of 2.5 µg/mL. (B) A confluent 10 cm dish of PH-Akt-GFP-expressing mouse embryonic fibroblasts (MEFs) is washed with PBS and treated with 0.05% trypsin-EDTA. The trypsin-treated cells are then split into a 15 mL centrifuge tube and a 6 cm tissue culture dish, both containing cell culture media. (C) From the 15 mL centrifuge tube, 500-1000 µL is pipetted onto the fibronectin-coated coverslip. (D) The 6 cm dish and the 35 mm dish with the coverslip containing the sparsely seeded cells are placed in a 37 °C incubator overnight. Once polarized, these cells will provide the frame of focus for the cell spreading acquisition. (E) An hour before image acquisition, the 6 cm dish's media is replaced with phenol-red free DMEM supplemented with HEPES and the drug of interest. After 1 hour, the cells are treated with 0.05% trypsin-EDTA and transferred to a 15 mL centrifuge tube (Tube A) containing the HEPES/drug-supplemented phenol red free DMEM. The cells in Tube A are then further diluted in another 15 mL centrifuge tube (Tube B), which is placed in the incubator for 45 minutes. (F) As the cells recover, the magnetic chamber is prepared from bottom to top: first the bottom plate is placed on a flat surface, then the coverslip with the polarized cells, the silicone gasket, the main body of the chamber, and finally the transparent cover are laid on top. (G) 1 mL of drug-supplemented phenol red free DMEM is pipetted into the magnetic chamber, which is then brought to the microscope stage. A CFI Plan Apo Lambda 60X Oil objective is selected for image acquisition. (H) The transparent cover is removed and 500 µL of Tube B's contents are pipetted into the magnetic chamber. (I) For image acquisition, appropriate fields of view will contain green "halos", which are suspended cells that have not yet attached to the coverslip. (J) The cells are imaged for 15 minutes. Please click here to view a larger version of this figure.

Figure 2: The effect of recovery time on cell spreading. Cells maintained in suspension for the indicated time (cell recovery step in the protocol) were plated on fibronectin-coated coverslips for 15 minutes, fixed with 4% paraformaldehyde and imaged by phase contrast microscopy. (A) Top panels: phase contrast images acquired with a 20X objective. Bottom panels: watershed-segmented cell masks with the cell areas color-coded. (B) Quantification of cell area. Please click here to view a larger version of this figure.

Figure 3: Graphical User Interface (GUI) and the working principles of image processing and analysis software. (A) The GUI of the "Cell spread area" tab. Refer to Step 5.3 for instructions. (B) The GUI of the "Kymograph generator & analysis" tab. Refer to Step 6 for instructions. (C) The image processing pipeline of the software. The software first identifies spreading cells (labeled by a green bounding box) in the whole field of view. The spreading cells are identified based on their intensity value, circularity, and aspect ratio. The identified spreading cells are then segmented frame-by-frame using global intensity thresholding. Each binary mask is processed by median filtering and binary hole filling followed by morphological closing to smoothen the cell edge. The red outline corresponds to the segmented cell boundary. The cell's area, aspect ratio, and circularity are extracted from the binary cell map. The graph shows the area and circularity of a representative cell over time. Upon cell seeding, cells do not start spreading immediately, giving rise to the lag phase seen on the graph. Following the lag phase, cells spread rapidly during the rapid expansion phase and eventually reach a plateau phase. Please click here to view a larger version of this figure.

Figure 4: Representative results of cell spread area analysis upon Arp2/3 inhibition. (A) Representative images of PH-Akt-GFP-expressing MEFs spreading on a fibronectin-coated coverslip over the course of 3 minutes. The red line indicates the cell boundary extracted by the cell segmentation algorithm. Top panels: 0.1% DMSO-treated cells. Bottom panels: 100 µM CK-666 (Arp2/3 inhibitor)-treated cells. (B) A graph showing cell spread area over time. The cell spread area was quantified as fold changes relative to the average cell spread area of control cells. Blue and pink lines represent control and Arp2/3-inhibited cells, respectively. The shaded regions indicate the upper and lower standard deviation of the cell spread area. (C) A bar plot with individual data points showing the average cell circularity of control and Arp2/3-inhibited cells. All error bars represent standard deviation. *, p < 0.05, **, p < 0.01, ***, p < 0.001, n.s. (not significant, p > 0.05) as detected by parametric student t-tests. Please click here to view a larger version of this figure.

Figure 5: Representative results of kymograph analysis of spreading cells upon Arp2/3 inhibition. (A – B) Representative images and kymographs extracted from 0.1% DMSO-treated (control) cells and 100 µM CK-666-treated (Arp2/3 inhibitor) cells. Left panels: inverted grayscale images of a control and Arp2/3-inhibited cell. The dashed lines correspond to the pre-defined lines where kymographs were extracted. Right panels: kymographs are extracted along the dashed lines shown on the grayscale images. The plot boundaries are color-coded to match the dashed lines on the grayscale images. The dashed line in each plot indicates the slope of the curve from which the average protrusion speed was calculated. To pinpoint the plateau phase, a logistic growth curve was fitted to the data points and the plateau was derived from the parameter, c (Supplementary Figure 1). The red dots denote the retraction events. (C) A bar plot with individual data points showing the average protrusion speeds of the control and Arp2/3-inhibited cells. All error bars represent standard deviation. (D) A bar plot with individual data points showing the frequency of retraction events of the control and Arp2/3-inhibited cells. (C – D) *, p < 0.05, **, p < 0.01, ***, p < 0.001, n.s. (not significant, p > 0.05) as detected by non-parametric Mann-Whitney tests. Please click here to view a larger version of this figure.
Supplemental Figure 1: A representative kymograph and the curve fitting result. A kymograph extracted from a spreading cell. The blue dashed line corresponds to the distance of the cell edge relative to the first frame. The red solid line corresponds to the fitted curve. To increase the confidence of the curve fitting, the raw data points were first smoothed by a Savitzky-Golay filter. After the curve-fitting, the parameter c, from the logistic equation, was used to identify the plateau point. The raw data point closest to c is designated as the plateau point. Please click here to download this File.
Supplemental Files. Please click here to download this File.
