The ability of bacteria to survive and thrive is critically dependent on how well they are able to perceive and respond to perturbations in their environments, and this in turn is dependent on their signal transduction systems. The number of signaling systems a bacterium encodes has been called its “microbial IQ” and can be an indication of both variability of its environment and its ability to sense multiple signals and fine tune its response1. Two component signal transduction systems (TCS) are the most prevalent signaling systems used by bacteria, and they consist of a histidine kinase (HK) that senses the external signal and transmits via phosphorylation to an effector response regulator (RR)2. RRs can have a variety of output domains and thus different effector modes, but the most common response is transcriptional regulation via a DNA binding domain1. The signals sensed and the corresponding functions of the vast majority of TCSs remain unknown.
Although in vivo methods such as ChIP-chip are routinely used for determination of genomic binding sites of transcription factors3, they can only be used for bacterial two component system RRs if the activating conditions or signals are known. Often the environmental cues that activate a TCS are harder to determine than their gene targets. The in vitro microarray based assay described here can be used to effectively and rapidly determine the gene targets and predict functions of TCSs. This assay takes advantage of the fact that RRs can be phosphorylated and thus activated in vitro using small molecule donors like acetyl phosphate4.
In this method, named DAP-chip for DNA-affinity-purified-chip (Figure 1), the RR gene of interest is cloned with a His-tag in E. coli, and a subsequently purified tagged protein is allowed to bind to sheared genomic DNA. The protein-bound DNA is then enriched by affinity-purification, the enriched and input DNA are amplified, fluorescently labeled, pooled together and hybridized to a tiling array that is custom made to the organism of interest (Figure 1). Microarray experiments are subject to artifacts and therefore additional steps are employed to optimize the assay. One such step is to attempt to determine one target for the RR under study using electrophoretic mobility shift assays (EMSA) (see workflow in Figure 2). Then, following binding to genomic DNA and the DAP steps, the protein-bound and input DNA are examined by qPCR to see if the positive target is enriched in the protein-bound fraction relative to the input fraction, thus confirming optimal binding conditions for the RR (Figure 2). After array hybridization, the data are analyzed to find peaks of higher intensity signal indicating genomic loci where the protein had bound. Functions may be predicted for the RR based on the gene targets obtained. The target genomic loci are used to predict binding site motifs, which are then experimentally validated using EMSAs (Figure 2). The functional predictions and gene targets for the RR may then be extended to closely related species that encode orthologous RRs by scanning those genomes for similar binding motifs (Figure 2). The DAP-chip method can provide a wealth of information for a TCS where previously there was none. The method can also be used for any transcriptional regulator if the protein can be purified and DNA binding conditions can be determined, and for any organism of interest with a genome sequence available.

Figure 1. The DNA-affinity-purified-chip (DAP-chip) strategy7. The RR gene from the organism of interest is cloned with a carboxy-terminal His-tag into an E. coli expression strain. Purified His-tagged protein is activated by phosphorylation with acetyl phosphate, and mixed with sheared genomic DNA. An aliquot of the binding reaction is saved as input DNA, while the rest is subjected to affinity purification using Ni-NTA resin. The input and the RR-bound DNA are whole genome amplified, and labeled with Cy3 and Cy5, respectively. The labeled DNA is pooled together and hybridized to a tiling array, which is then analyzed to determine the gene targets. Figure modified and reprinted using the creative commons license from7.

Figure 2. Summary of workflow. For any purified tagged protein, begin by determining a target using EMSA. Allow protein to bind genomic DNA and then DNA-affinity-purify (DAP) and whole genome amplify (WGA) the enriched and input DNA. If a gene target is known, use qPCR to ensure that the known target is enriched in the protein-bound fraction. If no target could be determined, proceed directly to DNA labeling and array hybridization. If enrichment by qPCR could not be observed, then repeat the protein-gDNA binding and DAP-WGA steps using different protein amounts. Use array analysis to find peaks and map them to target genes. Use the upstream regions of target genes to predict binding site motifs. Validate the motifs experimentally using EMSAs. Use the motif to scan the genomes of related species encoding orthologs of the RR under study, and predict genes targeted in those species as well. Based on the gene targets obtained, the physiological function of the RR and its orthologs may be predicted. Figure modified and reprinted using the creative commons license from7.
The above method was applied to determine the global gene targets of the RRs in the model sulfate reducing bacterium Desulfovibrio vulgaris Hildenborough7. This organism has a large number of TCSs represented by over 70 RRs, indicating the wide variety of possible signals that it senses and responds to. In vivo analyses on the functions of these signaling systems are hard to perform since their signals and thus their activating conditions are unknown. Here the DAP-chip method was used to determine the gene targets and thus predict possible functions for a representative RR DVU3023.
DVU3023 is a sigma-54-dependent RR encoded in an operon with its cognate HK (Figure 3A). The C-terminal His-tagged gene was cloned into and purified from E. coli. For initial target determination, the purified RR was tested for binding to its downstream operon which is a ten gene-operon (DVU3025-3035) consisting of lactate uptake and oxidation genes. RR DVU3023 shifted the upstream region of DVU3025 (Figure 3B). Next RR DVU3023 was allowed to bind to sheared D. vulgaris Hildenborough genomic DNA. Although phosphorylation was not required for binding to the promoter region of DVU3025, acetyl phosphate was added to the reaction in case it was required for binding to other promoters. Following affinity purification of the protein-bound DNA fraction, qPCR was used to show that the upstream region of DVU3025 is enriched (8.45 fold) in the protein-bound fraction (CT= 6.9) relative to the input DNA (Ct =9.98) (Figure 3C), thus indicating that the binding conditions used were appropriate for RR DVU3023. Lack of enrichment of the promoter region of a randomly chosen gene (DVU0013) was used as a negative control.
The protein-bound and input DNA samples were then labeled fluorescently and hybridized to a D. vulgaris Hildenborough tiling array that had a high probe density in the intergenic regions. The top four peaks were chosen as the most likely targets for DVU3023 (Figure 3D). These four peaks were followed by several others, which were also identified in DAP-chip analyses for several other RRs and hence appear to be sticky DNA (Table 1). Figure 3E is a schematic representation of the genes regulated by DVU3023. The positive target DVU3025 was the first peak obtained with the highest score. Two gene targets are two other singly encoded lactate permeases (DVU2451 and DVU3284). The fourth gene target does not lie in an upstream region, but in the intergenic region between two convergently transcribed genes/operons (DVU0652 and DVU0653). This is a large intergenic region, and additionally also encodes a predicted sigma54-dependent promoter. It is possible that there is an undiscovered sRNA encoded in this region that is regulated by RR DVU3023.
Using the upstream regions of the targets obtained by DAP-chip, MEME was used to predict a binding site motif (Figure 3F, Table 2). EMSA substrates carrying the specific motif upstream of DVU3025 were designed to confirm that RR DVU3023 recognizes and binds the predicted motif. The motif was further validated by making substitutions in the conserved bases within the motif which eliminated the binding shift (Figure 3G). This validated motif was then used in Perl-generated scripts to scan the genome sequences of other closely related sulfate reducing bacteria that had orthologs for DVU3023. Loci were chosen as possible gene targets when the motif was located in upstream regions of open reading frames (Table 3). Using the motif sequences predicted for the orthologous RRs, a consensus binding site motif was generated (Figure 3F) which closely resembled the one obtained for D. vulgaris Hildenborough alone.

Figure 3. Determining genomic targets for D. vulgaris response regulator DVU3023. A. DVU3023 is encoded in an operon with its cognate HK. The downstream operon has a sigma54-dependent promoter (bent black arrow) and was used as a candidate target gene. B. Purified RR DVU3023 bound and shifted the upstream region of DVU30257. C. q-PCR of upstream regions of DVU3025 (positive target) and DVU0013 (chosen as a negative control). E is protein-bound enriched DNA fraction, I is input DNA. D. Top four peaks obtained after DAP-chip analysis. Start and End refer to DNA coordinates at the start and end of the peaks; score refers to the log2R ratio of the fourth highest probe in the peak; Fdr = false discovery rate; cutoff_p is the cutoff percentage at which the peak was identified. E. Schematic representation of the gene targets for DVU3023 based on the DAP-chip peaks. Numbers in boxes indicate the peak number in D. The HK-RR genes are in green, target genes in blue and other genes in grey. Black bent arrows are sigma54-dependent promoters, green filled circles are predicted binding site motifs. Gene names are as follows: por –pyruvate ferredoxin oxidoreductase; llp – lactate permease; glcD- glycolate oxidase; glpC- Fe-S cluster binding protein; pta – phosphotransacetylase; ack- acetate kinase; lldE – lactate oxidase subunit; lldF/G – lactate oxidase subunit; MCP – methyl-accepting chemotaxis protein. F. Weblogo8 images of the predicted binding site motif. Top – derived from DAP-chip targets; Bottom – derived from binding sites present in genomes with orthologs of DVU30237. G. Validation of predicted binding site motif using EMSA. DVU3023 shifted the wild type motif (w) but not the modified motif (m). Sequences for the w and m motifs are shown on the right7. Figure modified and reprinted using the creative commons license from Rajeev et al7.

Table 1. Top 20 peaks from the DAP-chip array analysis. The table is divided into three sections. Peak attributes show details of the peaks as generated by array analysis software, where Location refers to whether the peak was found in the genome or the extrachromosomal plasmid, Start and End refer to the start and end loci for the peak, Score refers to the log2R ratios for the fourth highest probe in the peak, Fdr refers to the false discovery rate value, and cutoff_p is the cutoff percentage at which the peak was identified. The other two sections Start coordinate mapping and End coordinate mapping map the start and end loci, respectively, of the peak to the gene. In these sections Gene strand refers to the strand coding the gene, Offset indicates distance from the start of the gene (positive values indicate loci is upstream of gene, while negative values indicate loci is within the gene), Overlap gene value is TRUE if the locus overlaps a gene, DVU refers to the DVU# of the gene that the coordinates map to, and Description indicates the gene annotation. Table modified and reprinted using the creative commons license from Rajeev et al7. Please click here to view a larger version of this table.

Table 2. Sequences used to build the consensus DAP-chip target based motif in Figure 3. Table modified and reprinted using the creative commons license from Rajeev et al7.
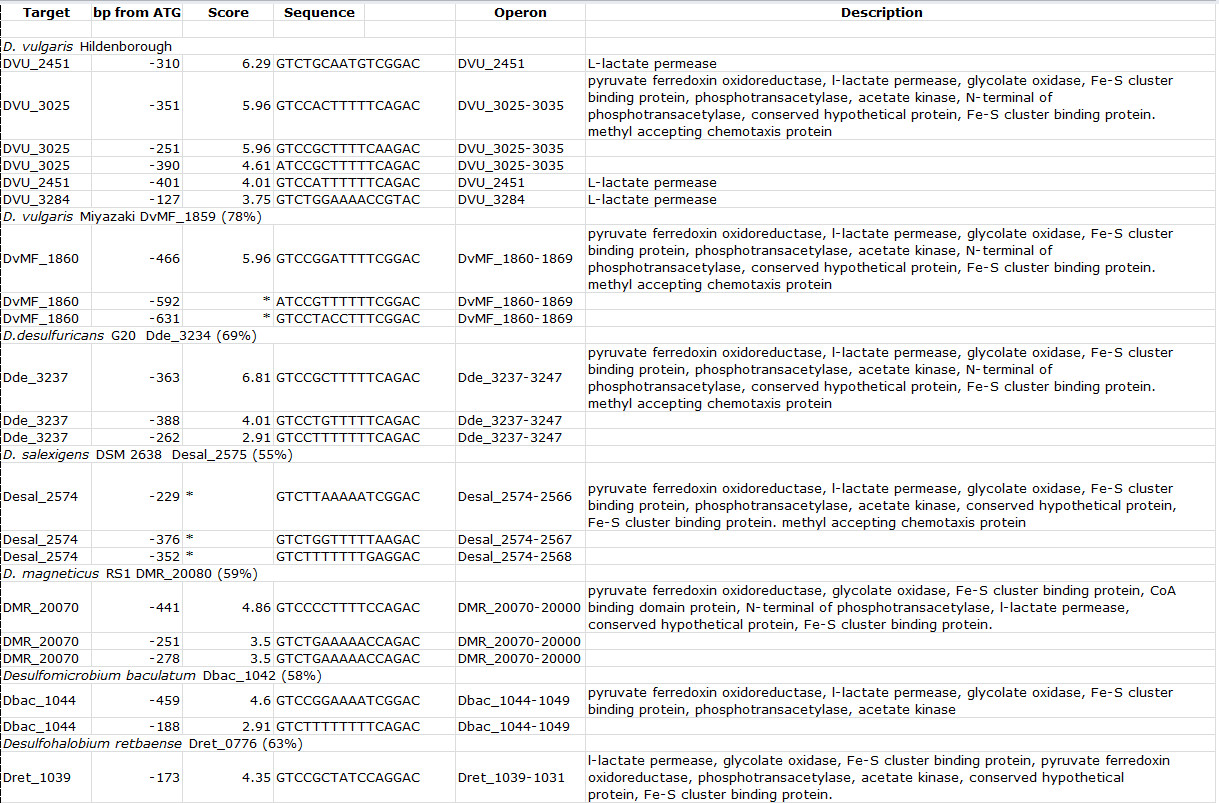
Table 3. Binding site motifs for DVU3023 orthologs present in other sequenced Desulfovibrio and related species. For each genome scanned, the organism name is indicated, followed by the locus tag for the DVU3023 ortholog and its percent identity to DVU3023 in parentheses. Score indicates the value assigned by the Perl program based on similarity to the input sequences. Description states the gene annotations for the genes in the target operon. Table modified and reprinted using the creative commons license from Rajeev et al7. Please click here to view a larger version of this table.
