



Overview
资料来源:埃瓦·布科夫斯卡-法尼班德1号,蒂尔德·安德斯森1号,罗尔夫·卢德1号
1临床科学隆德系,感染医学系,生物医学中心,隆德大学,221 00 隆德,瑞典
地球是数百万种细菌的栖息地,每种细菌物种都有其特殊特征。细菌物种的鉴定在微生物生态学中广泛用于确定环境样本的生物多样性和医学微生物学,以诊断受感染的患者。细菌可以使用传统的微生物学方法进行分类,如显微镜、特定介质的生长、生化和血清学测试以及抗生素敏感性检测。近几十年来,分子微生物学方法彻底改变了细菌鉴定。一个流行的方法是16S核糖体RNA(rRNA)基因测序。该方法不仅比传统方法更快、更准确,而且能够识别在实验室条件下难以生长的菌株。此外,在分子水平上对菌株进行分化,使表性相同的细菌(1-4)之间具有鉴别力。
16S rRNA 与 19 种蛋白质的复合物结合,形成细菌核糖体 (5) 的 30S 亚单位。它由16S rRNA基因编码,由于其在核糖体组装中的基本功能,在所有细菌中都存在并高度保存;然而,它也包含可变区域,可以作为特定物种的指纹。这些特征使16S rRNA基因成为理想的基因片段,可用于细菌的鉴定、比较和遗传分类(6)。
16S rRNA基因测序基于聚合酶链反应(PCR)(7-8),然后是DNA测序(9)。PCR 是一种分子生物学方法,用于通过一系列周期来扩增 DNA 的特定片段,其中包括:
i) 双绞合DNA模板的变性
ii) 与模板互补的引物(短寡核苷酸)的退火
iii) DNA聚合酶的引物延伸,合成新的DNA链
方法的原理图概述如图1所示。
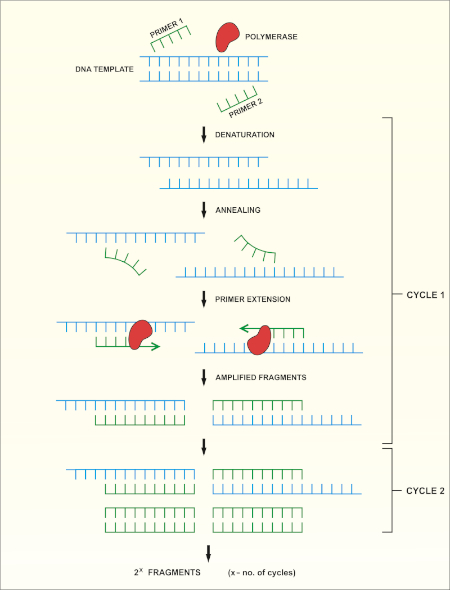
图 1:PCR 反应的原理概述。请点击此处查看此图的较大版本。
有几个因素对成功的PCR反应很重要,其中之一是DNA模板的质量。使用标准协议或商业试剂盒从细菌中分离染色体DNA。应特别注意获得不含污染物的DNA,这些污染物可以抑制PCR反应。
16S rRNA基因的保存区域允许设计通用引物对(一个正向和一个反向),可以结合和放大任何细菌物种的目标区域。目标区域的大小可能有所不同。虽然一些引热剂对可以扩增大多数16S rRNA基因,但其他仅扩增部分。常用引种示例如图1所示,其绑定位点如图2所示。
| 引种名称 | 序列 (5'~3') | 前进/后退 | 参考 |
| 8F b) | 阿加加特加特格格格格格格格股份公司 | 向前 | -1 |
| 27F | 阿加加特·加·加金茨格格格股份公司 | 向前 | -10 |
| 515F | 格格格格·CMGCCGGGTA | 向前 | -11 |
| 911R | 海合会加特克特科特加特加 | 反向 | -12 |
| 1391R | GACGGGGTGTGTRCA | 反向 | -11 |
| 1492R | GGTTACCTGTTT | 反向 | -11 |
表 1:用于扩增16S rRNA基因a)的标准寡核苷酸的例子。
a)通过计算正向和反向底漆的装订位点之间的距离(见图2),可以估计使用不同引漆组合生成的PCR产品的预期长度,例如PCR的大小使用底漆对 8F-1492R 的产品为 ±1500 bp,对于底漆对 27F-911R =900 bp。
b)也称为 fD1

图 2:16S rRNA序列和引结位点的代表性图。保留区域为灰色,可变区域用对角线填充。为了达到最高分辨率,引基8F和1492R(基于rRNA序列位置的名称)用于放大整个序列,从而允许对基因的几个可变区域进行测序。请点击此处查看此图的较大版本。
PCR 的循环条件(即脱氧核糖核酸变性、引物退火和合成所需的温度和时间)取决于所使用的聚合酶类型和引物的特性。建议遵循制造商针对特定聚合酶的指南。
PCR程序完成后,通过胶质电泳对产品进行分析。成功的 PCR 产生一个预期大小的单个波段。在测序之前,必须对产品进行纯化,以去除PCR反应中存在的残留底漆、脱氧核苷酸、聚合酶和缓冲液。纯化的DNA片段通常被发送到商业测序服务;然而,一些机构在他们自己的核心设施进行DNA测序。
DNA 序列由计算机从 DNA 色谱图自动生成,必须仔细检查质量,因为有时需要手动编辑。按照此步骤,将基因序列与沉积在16S rRNA数据库中的序列进行比较。识别相似区域,并传递最相似的序列。
Procedure
1. 设置
- 在处理微生物时,需要遵循良好的微生物实践。所有微生物,特别是未知样品,都应作为潜在的病原体处理。遵循无菌技术,避免污染样品、研究人员或实验室。处理细菌前后洗手,使用手套,并穿防护服。
- 对基因组DNA分离和PCR产品纯化的实验方案进行风险评估。有些试剂可能是有害的!
- 纯培养对16S rRNA测序至关重要。在继续分离基因组DNA之前,确保起始材料是完全纯净的。这可以通过条纹电镀来隔离单个菌落。如果需要,这些可以单独在盘子上或肉汤中进一步生长。
- 需要实验室设备:
- 用于 PCR 的热循环器。热循环器的功能是根据设定的程序提高和降低温度。创建程序时,系统将要求您输入每个 PCR 步骤的温度和时间值以及循环总数。
- 阿加玫瑰凝胶电泳系统。它用于根据DNA片段的大小和电荷分离DNA片段。在此协议中,阿加糖凝胶电泳将用于可视化分离基因组DNA和PCR产品的质量。
2. 议定书
注意:演示的规程适用于纯细菌培养的16S rRNA基因测序。它不适用于基因组学研究。
-
培养细菌以分离基因组DNA(gDNA)。
- 在合适的培养基上培养微生物。此步骤中可同时使用液体和固体介质。选择能产生最佳增长的条件。在计划实验时,请记住,缓慢生长的细菌可能需要几天才能达到晚点/静止生长阶段。在此协议中,在200rpm、37°C的摇动培养箱中一夜之间生长在乳原菌(LB)中。
-
gDNA 的隔离。
- 如果细菌生长在固体介质上,使用无菌循环刮擦一些细胞,并将其重新悬浮在1 mL的蒸馏水中
- 如果细菌在液体培养基中生长,则使用大约 1.5 mL 的隔夜培养液。
- 通过离心(1分钟,12,000 - 16,000 μ g)对细胞进行细胞细胞,去除上清液,并使用商用试剂盒或标准协议使用细胞进行gDNA分离[例如,CTAB总DNA制备(13)或苯酚氯仿提取(14)]。在这里,一个商业试剂盒用于从1.5 mL的B.subtilis 168过夜培养物中分离gDNA,OD600 = 1.5。
注1:对于一些革兰氏阴性细菌,这一步可以省略,并替换为简单的释放DNA从细胞沸腾。将细菌颗粒重新悬浮在蒸馏水中,并在100°C的加热块中孵育10分钟。
注2:革兰氏阳性细菌细胞难以破坏。因此,建议选择一种gDNA分离方法或试剂盒,专门用于从这组细菌中分离。
-
gDNA质量检查。
- 通过胶质胶电泳检查分离的gDNA的质量。首先,将5μL的分离gDNA与1μL的加载染料(6x)混合,并将样品加载到含有DNA染色试剂的0.8%角糖凝胶上。
- 加载分子质量标准并运行电泳,直到染料正面到达凝胶底部。
- 电泳完成后,将凝胶可视化到合适的透光器(紫外线或蓝光)。gDNA 显示为一个厚厚的高分子带(超过 10 kb)。gDNA质量检查的一个示例如图3所示。
- 如果gDNA通过质量控制(即存在高分子带,并且 gDNA 几乎没有涂抹),则通过首先将 3 个微离心管按如下标签进行稀释,如下所示:"10x"、"100x"和"1000x"。
- 将90μL的无菌蒸馏水放入3个管中的每一个。
- 取10μL的gDNA溶液,并将其添加到标有"10x"的管中。
- 彻底调入整个体积(即100μL),以确保溶液均匀混合。然后,从这个管子中拿出10μL的溶液,并将其转移到标有"100x"的管中。
- 如前所述混合,通过将溶液的 10 μL 从管"100x"转移到管"1000x"来重复相同的过程。这些稀释剂将用作PCR反应的模板。

图 3:从亚蒂利杆菌中分离出的gDNA的甘蔗糖凝胶电泳。车道 1:M - 分子质量标记(从上到下:10000 bp、8000 bp、6000 bp、5000 bp、4000 bp、3500 bp、3000 bp、2500 bp、2000 bp、1500 bp、1000 bp)。巷2:gDNA-基因组DNA从杆菌分离。请点击此处查看此图的较大版本。
-
PCR扩增16S rRNA基因。
注意:下面的PCR协议针对特定的DNA聚合酶和底漆对8F - 1492R进行了优化(见表1)。每个聚合酶和底漆对都需要优化该协议。- 解冻冰上的所有试剂。
- 准备 PCR 主混音,如表 2 所示。由于DNA聚合酶在室温下是活跃的,因此必须在冰上进行反应设置,即PCR管和反应成分应始终保存在冰上。为每个gDNA样品制备一个反应,为阴性对照准备一个反应。阴性对照是一种没有gDNA模板的PCR混合物,用于确保反应的其他成分不受污染。
注意:在多个样品的情况下,通常准备主组合。主混合是包含除模板之外的所有反应组件的解决方案。它有助于省略重复移液,避免移液错误,并确保样品之间的高度一致性。要准备主组合,请将每个组分(DNA模板除外)的体积乘以测试的样本数。混合微离心管中的所有组件,将整个体积上下调高几次。 - 主混合物的等分 49 μL 进入单个 PCR 管中。
- 将 1 μL 模板添加到带主混合的管中。对于负控制,添加 1 μL 无菌水。为确保组件充分混合,请用移液器设置为 30-50μL 轻轻移液 10 次。
- 使用表 3 所示的程序设置 PCR 机器。
- 将管放入 PCR 机器并启动程序。
- 程序完成后,通过胶质电泳检查PCR产品的质量。
- 使用 8F-1492R 底漆对成功的 PCR 反应产生大约 1.5 kb 的单波段(图 4)。如果存在其他频带(即非特定产品),则通过调整退火温度来优化 PCR 程序。如果存在预期大小的单个波段,则继续执行下一步。在这里,PCR反应与100倍稀释gDNA模板产生了最好的产品,因为它有一个锋利的带预期大小,缺乏非特异性产品。因此,它被选择被纯化并送去测序。
- 在测序之前,必须从PCR反应中存在的残留底漆、脱氧核苷酸、聚合酶和缓冲液中清除产品。PCR 产品可以使用商用 PCR 纯化套件进行隔离。PCR 反应加载在包含 DNA 结合基质的列上。PCR 产品绑定到列,而其他组件流经列。然后使用洗涤缓冲液洗涤柱,最后,DNA在选择的缓冲液中洗脱。确认随试剂盒补充的洗脱缓冲液与排序兼容。
- 发送纯化的PCR产物进行DNA测序。遵循在所选测序设施提交测序样本的准则。为了获得最佳序列覆盖率,请使用 PCR 扩增引子(与第 2.4.1 节中使用的相同)作为测序引子。此处,引注 8F 和 1492R 用于 PCR 产品测序。
| 组件 | 最终浓度 | 每个反应的体积 | 每 x 反应的体积(主混合) |
| 5x反应缓冲液 | 1x | 10 μL | 10 μL × x |
| 10 mM dNTP | 200 μM | 1 μL | 1 μL × x |
| 10 μM 底漆 8F | 0.5 μM | 2.5 μL | 2.5 μL × x |
| 10 μM 底漆 1492R | 0.5 μM | 2.5 μL | 2.5 μL × x |
| 磷聚合物酶 | 1 个单位 | 0.5 μL | 0.5 μL × x |
| 模板DNA | | - | 1 μL | - |
| ddH2O | - | 32.5 μL | 32.5 μL × x |
| 总体积 | 50 μL | 49 μL × x |
表 2:PCR 反应组分。• 使用步骤 2.3 中的 10 倍、100 倍或 1000x 稀释 gDNA。
| 步 | 温度 | 时间 | 周期 |
| 初始变性 | 98°C | 30 秒 | |
| 变性 | 98°C | 10 秒 | 25-30 |
| 退火 | 60°C | 30 秒 | |
| 扩展 | 72°C | 45 秒 | |
| 最终扩展 | 72°C | 7分钟 | |
| 保持 | 4°C | ∞ |
表 3:用于16S rRNA基因扩增的PCR程序。

图 4:以引体8F、1492R和gDNA为模板的PCR产物的Agarose凝胶电泳。B. subtilis的 gDNA 样本(见图 3)被稀释了 10、100 和 1000 次,以测试最佳结果。车道 1: M - 分子质量标记(从上到下:10000 bp、8000 bp、6000 bp、5000 bp、4000 bp、3500 bp、3000 bp、2500 bp、2500 bp、1500 bp、1500 bp、750 bp、500 bp、250 bp)。车道 2:PCR反应与10倍稀释模板。车道 3:PCR反应与100倍稀释模板。车道 4:PCR反应与1000x稀释模板。车道5:(C-) - 阴性对照(无DNA模板的反应)。请点击此处查看此图的较大版本。
3. 数据分析和结果
注意:PCR 产品使用正向(此处为 8F)和反向(此处为 1492R)引漆进行排序。因此,生成两组数据序列,一组用于正向,一组用于反向引注。对于每个序列,至少生成两种类型的文件:i) 包含 DNA 序列的文本文件;ii) DNA 色谱图,显示测序运行的质量。
- 对于正向底漆,打开色谱图,并仔细检查序列。质量序列的理想色谱图应具有均匀间距的峰值和很少或根本没有背景信号(图 5A)。
- 如果色谱图质量不高,应丢弃序列,或者根据以下内容修改序列文本文件:
- 整个色谱图中存在双峰表示存在多个 DNA 模板。如果细菌培养物不纯,情况可能就是如此。应丢弃此类序列 (图5B)。
- 由于同一位置存在不同颜色的峰值,可能会出现模棱两可的色谱图。最常见的错误之一是两个不同颜色的峰值位于同一位置,并且排序软件对基的分配不当(图5C)。手动更正任何分配错误的核苷酸,并在文本文件中对其进行编辑。
- 低分辨率色谱图可能导致"广泛峰",这通常会导致这些区域的核苷酸计数错误(图5D)。此错误难以纠正,因此不应将进一步对齐步骤中可能出现的不匹配视为可靠。
- 色谱图读数质量差,且存在多个峰值,通常出现在序列的 5' 和 3' 端。某些音序器软件会自动删除这些低质量片段(图5E),并且核苷酸不包括在文本文件中。如果序列未自动截断,请确定末端的低质量片段(例如弱信号、重叠峰值、分辨率损失),并从文本文件中删除相应的基。

图 5:DNA测序故障排除示例。A) 质量色谱图序列的示例(均匀间距、明确的峰值)。B) 质量较差的序列,通常发生在色谱图的开头。灰色区域被视为低质量,由排序软件自动删除。可以手动修剪更多基。C) 存在双峰(用箭头表示)。由红色箭头指示的核苷酸已被序列器读取为"T"(红色峰值),但蓝色峰值更强,也可以解释为"C"。D) 重叠峰表示DNA污染(即多个模板)。E) 分辨率损失和所谓的"宽峰"(以矩形标记),阻止可靠的基点调用。请点击此处查看此图的较大版本。
- 对反向底漆重复 3.1 和 3.2。
- 最后,将正向和反向序列组装成一个连续序列。良好的测序运行产生高达1100 bp的序列。 考虑到PCR产物长1500bp,使用正向和反向引漆获得的序列应部分重叠。
- 使用 DNA 序列组装程序合并两个序列,例如CAP3 (http://doua.prabi.fr/software/cap3) (15) 等免费工具。
- 将 FASTA 格式的两个序列插入到指示框中。单击"提交"按钮并等待结果返回。
- 要查看已组装的序列,请按结果选项卡中的"Contigs"。要查看对齐的详细信息,请按"装配详细信息"。
注1:如果使用 CAP3 软件进行相邻装配,则无需将反向引油序列转换为反向互补;但是,如果使用其他程序,则可能需要此步骤。
注2:FASTA 格式是一种基于文本的格式,用于表示核苷酸序列。FASTA 文件中的第一行(描述行)以符号">"开头,后跟序列的名称或唯一标识符。描述线之后是核苷酸序列。以以下格式粘贴序列:
>序列_frw_引基
在此处从文本文件粘贴序列
>序列_rvs_底漆
在此处从文本文件粘贴序列
- 通过访问基本本地对齐搜索工具(BLAST;https://blast.ncbi.nlm.nih.gov/Blast.cgi)的网站进行数据库搜索。
- 选择"核苷酸 BLAST"工具,将序列与数据库进行比较。
- 将序列(在 3.5 中组装的连续序列)输入"查询序列"文本框中,然后在向下滚动菜单中选择数据库"16S rRNA 序列(细菌和 Archea)。"。
- 按页面底部的"BLAST"按钮。将返回最相似的序列。图 6显示了一个示例 BLAST 结果。在提出的实验中,最热门的是B.subtilis菌株168,显示100%的标识与BLAST数据库中可用的序列。
- 如果热门命中未显示 100% 标识,请转到对齐路线并检查不匹配。单击顶部点击后,您将被定向到对齐的详细信息。对齐的核苷酸将由短的垂直线连接,而不匹配的核苷酸之间有间隙。返回到从测序公司收到的色谱图,并再次修改序列,重点是不匹配的区域。如果发现任何其他错误,请更正该序列。使用更正的序列再次运行 BLAST。

图 6:核苷酸BLAST结果示例。16S rRNA基因序列来自纯培养B.subtilis 168作为查询序列。命中顶部显示 100% 标识 (下划线) 到B. subtilis应变 168, 如预期.请点击此处查看此图的较大版本。
地球是数以百万计的细菌物种的家园,每个物种都有独特的特征。识别这些物种对于评估环境样本至关重要。医生还需要区分不同的细菌种类来诊断感染的病人。
为了识别细菌,可以使用多种技术,包括对特定介质上的形态或生长进行微观观察,以观察菌群形态。基因分析是另一种识别细菌的技术,近年来越来越受欢迎,部分是由于16S核糖体RNA基因测序。
细菌核糖体是一种蛋白质RNA复合物,由两个亚单位组成。30S 亚单位,这两个亚单位中较小的,包含 16S rRNA,由基因组 DNA 中包含的 16S rRNA 基因编码。16S rRNA 的特定区域由于在核糖体组装中的基本功能而高度保存。而其他地区,不太关键的功能,可能不同细菌物种。16S rRNA 中的可变区域可作为细菌物种的独特分子指纹,使我们能够区分表象相同的菌株。
获得gDNA质量样本后,16S rRNA编码基因的PCR可以开始。PCR是一种常用的分子生物学方法,包括双链DNA模板变性周期、通用引物对退火(扩增基因高度保守的区域)以及DNA聚合酶对引物的延伸。虽然一些引源扩增了大多数16S rRNA编码基因,但其他引体只放大了它的碎片。PCR后,可通过胶质胶电泳对产物进行分析。如果扩增成功,凝胶应包含一个预期大小的单带,具体取决于所使用的底漆对,最高为1500bp,即16S rRNA基因的近似长度。
经过纯化和测序,获得的序列可以输入BLAST数据库,与参考16S rRNA序列进行比较。由于此数据库基于最高相似性返回匹配,因此可以确认感兴趣的细菌的身份。在本视频中,您将观察 16S rRNA 基因测序,包括 PCR、DNA 序列分析和编辑、序列组装和数据库搜索。
在处理微生物时,必须遵循良好的微生物实践,包括使用无菌技术和穿戴适当的个人防护设备。对感兴趣的微生物或环境样本进行适当的风险评估后,获得测试培养物。在此示例中,使用了纯亚氏杆菌区域性。
首先,在适当的条件下,在合适的介质上生长微生物。在本例中,在LB肉汤中生长一夜,在37摄氏度的摇动培养箱中设置为200rpm。接下来,使用市售试剂盒从1.5毫升的B.subtilis过夜培养物中分离基因组DNA或gDNA。
为了检查分离的DNA的质量,首先将5微升的分离gDNA与一微升的DNA凝胶加载染料混合。然后,将样品加载到含有DNA染色试剂的0.8%角糖凝胶中,如SYBR安全剂或EtBr。在此之后,将一千基分子质量标准加载到凝胶上,并运行电泳,直到前染料距离凝胶底部约 0.5 厘米。凝胶电泳完成后,在蓝光转光器上可视化凝胶。gDNA 应显示为一个厚带,大小超过 10 千基,并且具有最小的涂抹。
之后,要创建 gDNA 的连续稀释,将三个微离心管标记为 10X、100X 和 1000X。然后,使用移液器将 90 微升无菌蒸馏水放入每个管中。接下来,在 10X 管中加入 10 微升的 gDNA 溶液。上下移移整个体积,以确保溶液彻底混合。然后,从 10X 管中取出 10 微升溶液,并将其转移到 100X 管中。混合解决方案,如前所述。最后,将10微升溶液在100X管中转移到1000X管中。
要开始 PCR 协议,在冰上解冻必要的试剂。然后,准备 PCR 主组合。由于DNA聚合酶在室温下是活跃的,因此反应必须发生在冰上。将49微升的主混合物混合到每个PCR管中。然后,在每个实验管中加入一个微升模板,在负控制管中加入一微升无菌水,上下移液混合。在此之后,根据表中描述的程序设置 PCR 机器。将管子放入热循环器中,然后启动程序。
程序完成后,通过胶质电泳检查产品质量,如前所述。使用所述协议的成功反应应产生大约 1.5 千碱的单个波段。在此示例中,含有 100X 稀释 gDNA 的样品产生了最高质量的产品。接下来,用市售的试剂盒来纯化最好的PCR产品,在本例中为100X gDNA。现在,PCR 产品可以发送进行测序。
在此示例中,PCR 产品使用正向和反向引漆进行排序。因此,生成两个数据集,每个数据集包含 DNA 序列和 DNA 色谱图:一个用于正向引体,另一个用于反向引色。首先,检查从每个引漆生成的色谱图。理想的色谱图应具有均匀间距的峰值,几乎没有背景信号。
如果色谱图显示双峰,PCR 产品中可能存在多个 DNA 模板,应丢弃序列。如果色谱图在同一位置包含不同颜色的峰值,则测序软件可能误称核苷酸。可以在文本文件中手动识别和更正此错误。色谱中存在宽峰表示分辨率损失,导致相关区域的核苷酸计数错误。此错误很难更正,任何后续步骤中的不匹配都应被视为不可靠。色谱图读数质量差,由存在多个峰值指示,通常发生在序列的五个素数和三个素端。某些排序程序会自动删除这些低质量部分。如果序列未自动截断,请识别低质量片段,并从文本文件中删除各自的基。
使用 DNA 组装程序将两个引体序列组装成一个连续序列。请记住,使用正向和反向引漆获得的序列应部分重叠。在 DNA 装配程序中,以 FASTA 格式将两个序列插入相应的框中。然后,单击提交按钮并等待程序返回结果。
要查看已装配的序列,请单击结果选项卡中的"Contigs"。然后,要查看对齐的详细信息,请选择装配体详细信息。导航到网站的基本本地对齐搜索工具,或BLAST,并选择核苷酸BLAST工具,以比较您的序列与数据库。在查询序列文本框中输入序列,并在向下滚动菜单中选择相应的数据库。最后,单击页面底部的 BLAST 按钮,然后等待该工具从数据库中返回最相似的序列。
在此示例中,命中数最高的是B. subtilis应变 168,显示与 BLAST 数据库中序列的 100% 标识。如果热门命中未显示您预期物种或应变的 100% 标识,请单击与您的查询最匹配的序列以查看对齐的详细信息。对齐的核苷酸将由短的垂直线连接,不匹配的核苷酸之间会有间隙。聚焦已识别的不匹配区域,修改序列并根据需要重复 BLAST 搜索。
Subscription Required. Please recommend JoVE to your librarian.
Applications and Summary
识别细菌物种对于不同的研究人员以及医疗保健人员都很重要。16S rRNA测序最初被研究人员用于确定细菌之间的遗传关系。随着时间的推移,它已被实施在基因组学研究中,以确定环境样本的生物多样性和临床实验室作为一种识别潜在病原体的方法。它能够快速准确地识别临床样本中的细菌,促进早期诊断和加快患者治疗。
Subscription Required. Please recommend JoVE to your librarian.
References
- Weisburg, W.G., Barns, S.M., Pelletier, D.A. and Lane D.J. 16S ribosomal DNA amplification for phylogenetic study. J Bacteriol. 173 (2): 697-703. (1991)
- Drancourt, M., Bollet, C., Carlioz, A., Martelin, R., Gayral, J.P., Raoult D. 16S ribosomal DNA sequence analysis of a large collection of environmental and clinical unidentifiable bacterial isolates. J Clin Microbiol. 38 (10):3623-3630. (2000)
- Woo, P.C., Lau, S.K., Teng, J.L., Tse, H., Yuen, K.Y. Then and now: use of 16S rDNA gene sequencing for bacterial identification and discovery of novel bacteria in clinical microbiology laboratories. Clin Microbiol Infect. 14 (10):908-934. (2008)
- Tang, Y.W., Ellis, N.M., Hopkins, M.K., Smith, D.H., Dodge, D.E., Persing, D.H. Comparison of phenotypic and genotypic techniques for identification of unusual aerobic pathogenic gram-negative bacilli. J Clin Microbiol. 36 (12):3674-3679. (1998)
- Tsiboli, P., Herfurth, E., Choli, T. Purification and characterization of the 30S ribosomal proteins from the bacterium Thermus thermophilus. Eur J Biochem. 226 (1):169-177. (1994)
- Woese, C.R. Bacterial evolution. Microbiol Rev. 51 (2):221-271. (1987)
- Bartlett, J.M., Stirling, D. A short history of the polymerase chain reaction. Methods Mol Biol. 226:3-6. (2003)
- Wilson, K.H., Blitchington, R.B., Greene, R.C. Amplification of bacterial 16S ribosomal DNA with polymerase chain reaction. J Clin Microbiol. 28 (9):1942-1946. (1990)
- Shendure, J., Balasubramanian, S., Church, G.M., Gilbert, W., Rogers, J., Schloss, J.A., Waterston, R.H. (2017) DNA sequencing at 40: past, present and future. Nature. 550:345-353.
- Lane, D.J. 16S/23S rRNA sequencing. (1991) In Nucleic acid techniques in bacterial systematics. (Goodfellow, M. and Stackebrandt, E., eds.) p.115-175. Wiley and Sons, Chichester, United Kingdom.
- Turner, S., Pryer, K.M., Miao, V.P., Palmer, J.D. (1999) Investigating deep phylogenetic relationships among cyanobacteria and plastids by small subunit rRNA sequence analysis. J Eukaryot Microbiol. 46:327-338.
- Fredricks, D.N., Relman, D.A. (1998) Improved amplification of microbial DNA from blood cultures by removal of the PCR inhibitor sodium polyanetholesulfonate. J Clin Microbiol. 36:2810-2816.
- Wilson, K. Preparation of genomic DNA from bacteria. (2001) Curr Protoc Mol Biol. Chapter 2:Unit 2.4.
- Wright, M. H., Adelskov, J., Greene, A.C. (2017) Bacterial DNA extraction using individual enzymes and phenol/chloroform separation. J Microbiol Biol Educ. 18:18.2.48.
- Huang, X., Madan, A. (1999). CAP3: A DNA sequence assembly program. Genome Res. 9:868-877.