Earth is home to millions of bacterial species, each with unique characteristics. Identifying these species is critical in evaluating environmental samples. Doctors also need to distinguish different bacterial species to diagnose infected patients.
To identify bacteria, a variety of techniques can be employed, including microscopic observation of morphology or growth on a specific media to observe colony morphology. Genetic analysis, another technique for identifying bacteria has grown in popularity in recent years, due in part to 16S ribosomal RNA gene sequencing.
The bacterial ribosome is a protein RNA complex consisting of two subunits. The 30S subunit, the smaller of these two subunits, contains 16S rRNA, which is encoded by the 16S rRNA gene contained within the genomic DNA. Specific regions of 16S rRNA are highly conserved, due to their essential function in ribosome assembly. While other regions, less critical to function, may vary among bacterial species. The variable regions in 16S rRNA, can serve as unique molecular fingerprints for bacterial species, allowing us to distinguish phenotypically identical strains.
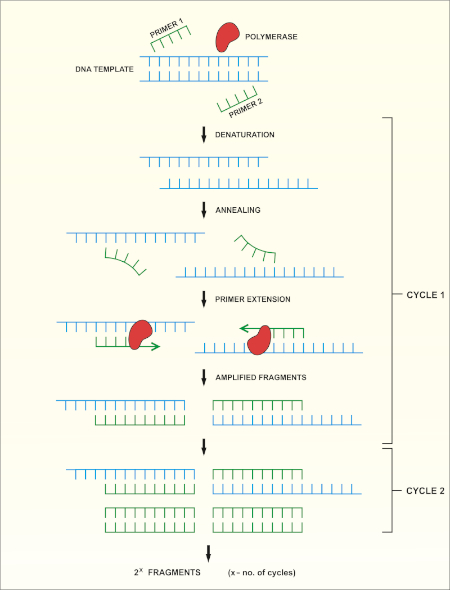
After obtaining a quality sample of gDNA, PCR of the 16S rRNA encoding gene can begin. PCR is a commonly used molecular biology method, consisting of cycles of denaturation of the double-stranded DNA template, annealing of universal primer pairs, which amplify highly conserved regions of the gene, and the extension of primers by DNA polymerase. While some primers amplify most of the 16S rRNA encoding gene, others only amplify fragments of it. After PCR, the products can be analyzed via agarose gel electrophoresis. If amplification was successful, the gel should contain a single band of an expected size, depending upon the primer pair used, up to 1500bp, the approximate length of the 16S rRNA gene.
After purification and sequencing, the obtained sequences can then be entered into the BLAST database, where they can be compared with reference 16S rRNA sequences. As this database returns matches based on the highest similarity, this allows confirmation of the identity of the bacteria of interest. In this video, you will observe 16S rRNA gene sequencing, including PCR, DNA sequence analysis and editing, sequence assembly and database searching.
When handling microorganisms, it is essential to follow good microbiological practice, including using aseptic technique and wearing appropriate personal protective equipment. After performing an appropriate risk assessment for the microorganism or environmental sample of interest, obtain a test culture. In this example, a pure culture of Bacillus subtilis is used.
To begin, grow your microorganism on a suitable medium in the appropriate conditions. In this example, Bacillus subtilis 168 is grown in LB broth overnight in a shaking incubator set to 200 rpm at 37 degrees Celsius. Next, use a commercially available kit to isolate genomic DNA or gDNA from 1.5 milliliters of the B. subtilis overnight culture.
To check the quality of the isolated DNA, first mix five microliters of the isolated gDNA with one microliter of DNA gel loading dye. Then, load the sample onto a 0.8% agarose gel, containing DNA staining reagent, such as SYBR safe or EtBr. After this, load a one kilobase molecular mass standard onto the gel, and run the electrophoresis until the front dye is approximately 0.5 centimeters from the bottom of the gel. Once the gel electrophoresis is complete, visualize the gel on a blue light transilluminator. The gDNA should appear as a thick band, above 10 kilobase in size and have minimal smearing.
After this, to create serial dilutions of the gDNA, label three microcentrifuge tubes as 10X, 100X, and 1000X. Then, use a pipette to dispense 90 microliters of sterile distilled water into each of the tubes. Next, add 10 microliters of the gDNA solution to the 10X tube. Pipette the whole volume up and down to ensure the solution is mixed thoroughly. Then, remove 10 microliters of the solution from the 10X tube and transfer this to the 100X tube. Mix the solution as previously described. Finally, transfer 10 microliters of the solution in the 100X tube, to the 1000X tube.
To begin the PCR protocol, thaw the necessary reagents on ice. Then, prepare the PCR master mix. Since the DNA polymerase is active at room temperature, the reaction set up must occur on ice. Aliquot 49 microliters of the master mix into each of the PCR tubes. Then, add one microliter of template to each of the experimental tubes and one microliter of sterile water to the negative control tube, pipetting up and down to mix. After this, set the PCR machine according to the program described in the table. Place the tubes into the thermocycler and start the program.
Once the program is complete, examine the quality of your product via agarose gel electrophoresis, as previously demonstrated. A successful reaction using the described protocol should yield a single band of approximately 1.5 kilobase. In this example, the sample containing 100X diluted gDNA yielded the highest quality product. Next, purify the best PCR product, in this case, the 100X gDNA, with a commercially available kit. Now the PCR product can be sent for sequencing.
In this example, the PCR product is sequenced using forward and reverse primers. Thus, two data sets, each containing a DNA sequence and a DNA chromatogram, are generated: one for the forward primer and the other for the reverse primer. First, examine the chromatograms generated from each primer. An ideal chromatogram should have evenly spaced peaks with little to no background signals.
If the chromatograms display double peaks, multiple DNA templates may have been present in the PCR products and the sequence should be discarded. If the chromatograms contained peaks of different colors in the same location, the sequencing software likely miscalled nucleotides. This error can be manually identified and corrected in the text file. The presence of broad peaks in the chromatogram indicates a loss of resolution, which causes miscounting of the nucleotides in the associated regions. This error is difficult to correct and mismatches in any of the subsequent steps should be treated as unreliable. Poor chromatogram reading quality, indicated by the presence of multiple peaks, usually occurs at the five prime and three prime ends of the sequence. Some sequencing programs remove these low quality sections automatically. If your sequence was not truncated automatically, identify the low quality fragments and remove their respective bases from the text file.
Use a DNA assembly program to assemble the two primer sequences into one continuous sequence. Remember, sequences obtained using forward and reverse primers should partially overlap. In the DNA assembly program, insert the two sequences in FASTA format into the appropriate box. Then, click the submit button and wait for the program to return the results.
To view the assembled sequence, click on Contigs in the results tab. Then, to view the details of the alignment, select assembly details. Navigate to the website for the basic local alignment search tool, or BLAST, and select the nucleotide BLAST tool to compare your sequence to the database. Enter your sequence into the query sequence text box and select the appropriate database in the scroll down menu. Finally, click the BLAST button on the bottom of the page, and wait for the tool to return the most similar sequences from the database.
In this example, the top hit is B. subtilis strain 168, showing 100% identity with the sequence in the BLAST database. If the top hit does not show 100% identity to your expected species or strain, click on the sequence which most closely matches your query to see the details of the alignment. Aligned nucleotides will be joined by short vertical lines and mismatched nucleotides will have gaps between them. Focusing on the identified mismatched regions, revise the sequence and repeat the BLAST search if desired.