Quelle: Ewa Bukowska-Faniband1, Tilde Andersson1, Rolf Lood1
1 Institut für Klinische Wissenschaften Lund, Abteilung für Infektionsmedizin, Biomedical Center, Universität Lund, 221 00 Lund, Schweden
Planet Erde ist ein Lebensraum für Millionen von Bakterienarten, von denen jede spezifische Eigenschaften hat. Die Identifizierung von Bakterienarten wird in der mikrobiellen Ökologie häufig verwendet, um die Artenvielfalt von Umweltproben und medizinische Mikrobiologie zur Diagnose infizierter Patienten zu bestimmen. Bakterien können mit konventionellen mikrobiologischen Methoden wie Mikroskopie, Wachstum auf bestimmten Medien, biochemischen und serologischen Tests und Antibiotika-Empfindlichkeitstests klassifiziert werden. In den letzten Jahrzehnten haben molekulare mikrobiologische Methoden die bakterielle Identifizierung revolutioniert. Eine beliebte Methode ist die 16S ribosomale RNA (rRNA) Gensequenzierung. Diese Methode ist nicht nur schneller und genauer als herkömmliche Methoden, sondern ermöglicht auch die Identifizierung von Stämmen, die unter Laborbedingungen schwer zu züchten sind. Darüber hinaus ermöglicht die Differenzierung von Stämmen auf molekularer Ebene eine Diskriminierung von phänotypisch identischen Bakterien (1-4).
16S rRNA verbindet sich mit einem Komplex von 19 Proteinen zu einer 30S-Untereinheit des bakteriellen Ribosoms (5). Es wird durch das 16S rRNA-Gen kodiert, das in allen Bakterien aufgrund seiner wesentlichen Funktion in der Ribosom-Montage vorhanden und hochkonserviert ist; Es enthält jedoch auch variable Regionen, die als Fingerabdrücke für bestimmte Arten dienen können. Diese Eigenschaften haben das 16S rRNA-Gen zu einem idealen genetischen Fragment gemacht, das bei der Identifizierung, dem Vergleich und der phylogenetischen Klassifizierung von Bakterien verwendet werden kann (6).
16S rRNA Gensequenzierung basiert auf der Polymerase-Kettenreaktion (PCR) (7-8) gefolgt von DNA-Sequenzierung (9). PCR ist eine molekularbiologische Methode, die verwendet wird, um bestimmte DNA-Fragmente durch eine Reihe von Zyklen zu verstärken, die Folgendes umfassen:
i) Denaturierung einer doppelsträngigen DNA-Vorlage
ii) Glühen von Primern (kurze Oligonukleotide), die die Vorlage ergänzen
iii) Erweiterung der Primer durch das DNA-Polymerase-Enzym, das einen neuen DNA-Strang synthetisiert
Eine schematische Übersicht über die Methode ist in Abbildung 1dargestellt.
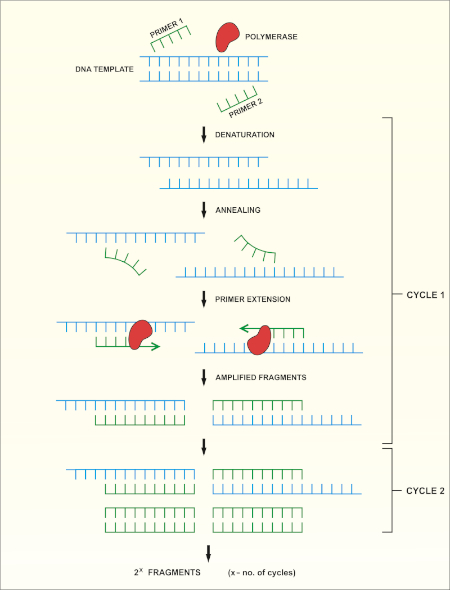
Abbildung 1: Schematische Übersicht über die PCR-Reaktion. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
Es gibt mehrere Faktoren, die für eine erfolgreiche PCR-Reaktion wichtig sind, einer davon ist die Qualität der DNA-Vorlage. Die Isolierung chromosomaler DNA von Bakterien kann mit Standardprotokollen oder kommerziellen Kits durchgeführt werden. Besondere Vorsicht sollte darauf geachtet werden, DNA zu erhalten, die frei von Verunreinigungen ist, die die PCR-Reaktion hemmen können.
Konservierte Regionen des 16S rRNA-Gens ermöglichen das Design universeller Primerpaare (ein Vorwärts- und ein Rückwärtsgang), die die Zielregion in jeder Bakterienart binden und verstärken können. Die Zielregion kann in ihrer Größe variieren. Während einige Primerpaare den größten Teil des 16S rRNA-Gens verstärken können, verstärken andere nur Teile davon. Beispiele für häufig verwendete Primer sind in Tabelle 1 dargestellt und ihre Bindungsstellen sind in Abbildung 2dargestellt.
| Primername | Sequenz (5”3′) | Vorwärts/ Rückwärts | verweis |
| 8F b) | AGAGTGTGATCCTGGCTCAG | vorwärts | -1 |
| 27F | AGAGTGTGATCMTGGCTCAG | vorwärts | -10 |
| 515F | GTGCCAGCMGCCGCGGTAA | vorwärts | -11 |
| 911R | GCCCCCGTCAATTCMTGA | Rückwärts | -12 |
| 1391R | GACGGGCGGTGTGTRCA | Rückwärts | -11 |
| 1492R | GGTTTTTTTACGACTT | Rückwärts | -11 |
Tabelle 1: Beispiele für Standardoligonukleotide, die bei der Amplifikation von 16S rRNA-Genen verwendet werden a).
a) Die erwarteten Längen des PCR-Produkts, das mit den verschiedenen Primerkombinationen erzeugt wird, können durch Berechnung des Abstands zwischen den Bindungsstellen für den Vorwärts- und den Reverse-Primer geschätzt werden (siehe Abbildung 2), z. B. die Größe der PCR Produkt mit Primer-Paar 8F-1492R ist 1500 bp, und für Primer-Paar 27F-911R 900 bp.
b) auch bekannt als fD1

Abbildung 2: Repräsentative Abbildung der 16S rRNA-Sequenz und der Primer-Bindungsstellen. Konservierte Bereiche sind grau gefärbt und variable Bereiche sind mit diagonalen Linien gefüllt. Um die höchste Auflösung zu ermöglichen, werden Primer 8F und 1492R (Name basierend auf der Position auf der rRNA-Sequenz) verwendet, um die gesamte Sequenz zu verstärken, was die Sequenzierung mehrerer variabler Bereiche des Gens ermöglicht. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
Die Fahrradbedingungen für PCR(d. h. die Temperatur und Die Zeit, die für die Denaturierung, das Geglüht mit Primern und synthetisiert erforderlich sind) hängen von der Verwendeten-Polymerase-Art und den Eigenschaften der Primer ab. Es wird empfohlen, die Herstellerrichtlinien für eine bestimmte Polymerase zu befolgen.
Nach Abschluss des PCR-Programms werden die Produkte durch Agarose-Gel-Elektrophorese analysiert. Eine erfolgreiche PCR ergibt ein einzelnes Band in der erwarteten Größe. Das Produkt muss vor der Sequenzierung gereinigt werden, um Restgrundierungen, Desoxyribonukleotide, Polymerase und Puffer zu entfernen, die in der PCR-Reaktion vorhanden waren. Die gereinigten DNA-Fragmente werden in der Regel zur Sequenzierung an kommerzielle Sequenzierungsdienste gesendet; Einige Institutionen führen jedoch DNA-Sequenzierungen in ihren eigenen Kerneinrichtungen durch.
Die DNA-Sequenz wird automatisch aus einem DNA-Chromatogramm von einem Computer erzeugt und muss sorgfältig auf Qualität überprüft werden, da manchmal eine manuelle Bearbeitung erforderlich ist. Nach diesem Schritt wird die Gensequenz mit Sequenzen verglichen, die in der 16S rRNA-Datenbank abgelagert sind. Die Regionen mit Ähnlichkeit werden identifiziert, und die ähnlichsten Sequenzen werden geliefert.