



Overview
Source: Andrew J. Van Alst1, Rhiannon M. LeVeque1, Natalia Martin1, et Victor J. DiRita1
1 Department of Microbiology and Molecular Genetics, Michigan State University, East Lansing, Michigan, États-Unis d'Amérique
Les courbes de croissance fournissent des informations précieuses sur la cinétique de croissance bactérienne et la physiologie cellulaire. Ils nous permettent de déterminer comment les bactéries réagissent dans des conditions de croissance variables ainsi que de définir des paramètres de croissance optimaux pour une bactérie donnée. Une courbe de croissance archétypale progresse à travers quatre étapes de croissance : lag, exponentiel, stationnaire, et la mort (1).
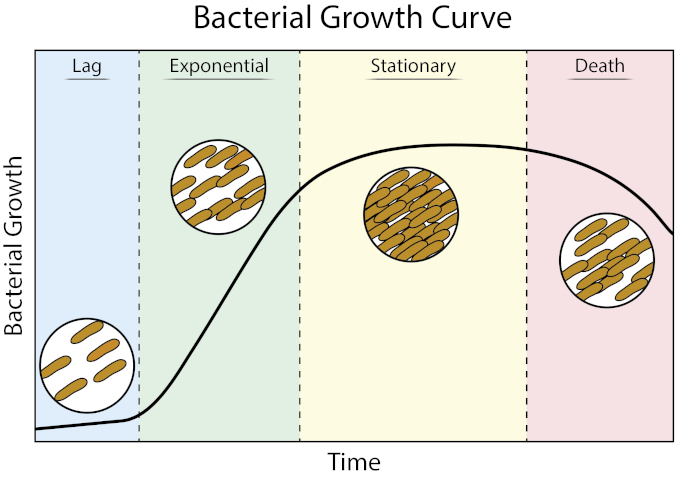
Figure 1 : Courbe de croissance bactérienne. Les bactéries cultivées dans la culture des lots progressent à travers quatre phases de croissance : lag, exponentiel, stationnaire, et la mort. La phase de décalage est la période de temps qu'il faut pour que les bactéries atteignent un état physiologique capable de croissance et de division cellulaires rapides. La phase exponentielle est l'étape de la croissance et de la division cellulaires les plus rapides au cours de laquelle la réplication de l'ADN, la transcription de l'ARN et la production de protéines se produisent à un rythme constant et rapide. La phase stationnaire se caractérise par un ralentissement et un plafonnement de la croissance bactérienne en raison de la limitation des nutriments et/ou de l'accumulation intermédiaire toxique. La phase de mort est l'étape au cours de laquelle la lyse cellulaire se produit à la suite d'une limitation nutritionnelle sévère.
La phase de décalage est la période de temps qu'il faut pour que les bactéries atteignent un état physiologique capable de croissance et de division cellulaires rapides. Ce décalage se produit parce qu'il faut du temps pour que les bactéries s'adaptent à leur nouvel environnement. Une fois que les composants cellulaires nécessaires sont générés en phase de décalage, les bactéries entrent dans la phase exponentielle de croissance où la réplication de l'ADN, la transcription d'ARN et la production de protéines se produisent
Procedure
1. Mise en place
- Matériaux de laboratoire requis : supports liquides, supports en agar solidifiés, flacons Erlenmeyer, tubes à essai de 15 ml, salin tamponné par phosphate (PBS), épandeur de cellules bactériennes, éthanol à 70 % et spectrophotomètre. Toutes les solutions et verrerie doivent être stérilisées avant utilisation.
- Préparer le poste de travail en stérilisant avec 70% d'éthanol. Travaillez près d'un brûleur Bunsen pour prévenir la contamination des médias.
- Lorsque vous travaillez avec des bactéries, un équipement de protection individuelle approprié et une technique aseptique doivent être utilisés. Une blouse de laboratoire et des gants sont nécessaires lorsque vous travaillez avec des cultures bactériennes.
- Recettes pour tampons, solutions et réactifs
- Saline tamponnée de phosphate (PBS) (8).
- Luria-Bertani Broth (LB) (9).
2. Protocole
- Préparation des médias
- Identifiez le support de croissance avec lequel développer les bactéries et préparez à la fois le bouillon liquide et l'agar solide (1,5% w/v agar) dans des bouteilles autoclavables séparées. Ici, bouillon LB et agar LB ont été préparés pour la croissance d'Escherichia coli.
- Stériliser les supports avec un bouchon semi-serré dans un autoclave réglé à 121 oC pendant 35 min.
- Pour les supports d'agar, après l'autoclage, placer dans un bain d'eau réglé à 50 oC pendant 30 minutes pour refroidir. Une fois refroidi, verser 20-25 ml de support d'agar dans des plats Petri circulaires de 100x15 mm. Laisser les assiettes être fixées 24 heures à température ambiante avant de les utiliser.
- Préparation initiale des bactéries
- À partir de bouillons congelés, les bactéries stries pour l'isolement sur l'agar média sélectionné pour obtenir des isolats de colonie unique. Incuber dans des conditions de croissance permises pour les bactéries choisies. Ici, E. coli est strié sur l'agar LB et est incubé à 37 oC pendant la nuit (16-18h).
- À l'aide d'une boucle d'inoculation stérile, sélectionnez une seule colonie à partir de la plaque de stries et inoculez 4 ml de support liquide dans un tube à essai de 15 ml et développez dans des conditions permises pour les bactéries choisies. Ici, E. coli est cultivé à 37 oC avec des secousses à 210 tr/min pendant la nuit (16-18h).
- Configuration de la courbe de croissance
- Préparation de flacon de croissance
- Autoclave flacons Erlenmeyer de taille appropriée. Typiquement, un rapport de 1:5 de médias au volume total de flacon est utilisé. Ici, un support LB de 100 ml est utilisé dans un flacon de 500 ml.
- À l'aide d'une pipette sérologique, transférer les supports stériles sur le flacon Erlenmeyer.
- Préparation de la série de dilution
- Étiqueter les tubes à essai de 15 mL : -1, -2, -3, -4, -5, -6, -7, -8 et -9, en distribuant 9mL PBS dans chacun. Ces chiffres correspondent au facteur de dilution utilisé pour calculer l'UFC/mL. Un nouvel ensemble de tubes est nécessaire pour chaque point de collecte. (Figure 2)
- Préparation de plaque d'agar
- Étiquette des plaques avec le temps de collecte et le facteur de dilution. Pour chaque point de temps, il y aura une plaque pour chaque dilution.
- Préparation de flacon de croissance
- Protocole de courbe de croissance
- Inoculation des médias
- En utilisant la culture liquide de nuit préparée dans le cadre de l'étape 2.2.2, inoculer les médias flasques avec 1:1000 volume de culture. Ici, la culture liquide de nuit de 100 l est ajoutée aux supports LB de 100 ml.
- Faites tourbillonner les médias pour répartir uniformément les bactéries.
- Collection Timepoint
- Configuration de l'état de croissance
- Placez le flacon dans des conditions de croissance expérimentales choisies pour les bactéries données. Les points temporels doivent être pris fréquemment pour les bactéries à croissance rapide et peuvent être pris à intervalles plus longs pour les bactéries à croissance lente. Ici, E. coli est cultivé à 37oC avec des secousses à 210 tours par minute (rpm) et des chronopoints pris toutes les 1 heure.
- Mesure de la densité optique (OD600)
- À chaque point de temps, y compris le point de départ (t ' 0), retirez 1 ml de culture bactérienne et distribuez dans une cuvette spectrophotomètre.
- Essuyez la cuvette propre et enregistrez la densité optique à 600 nm longueur d'onde. Si la lecture de densité optique est supérieure à 1,0, diluer 100 L de culture 1:10 avec 900 'L de nouveaux médias, enregistrer la densité optique, et multiplier cette valeur par 10 pour la mesure OD600.
- Mesure de l'unité de formation de colonies (CFU/mL)
- À chaque moment, retirer 1 ml de culture bactérienne et de distribuer dans le tube à essai en verre -1 contenant 9 ml de PBS.
- Pour la série de dilution, transférer en série 1 mL du tube -1 vers le bas tous les tubes de dilution à la -9, vortexing après chaque transfert. (Figure 2)
- Pour chaque dilution, distribuez 100 l de suspension cellulaire à la plaque d'agar solide de médias étiquetée en conséquence. (Figure 2)
- À l'aide d'un épandeur cellulaire qui a été stérilisé dans l'éthanol, passé à travers une flamme de brûleur Bunsen, et refroidi en touchant la surface de l'agar, répandre les 100 L de suspension cellulaire jusqu'à ce que la surface de la plaque d'agar devient sèche.
- Incuber les plaques de propagation à l'envers à une température qui favorise la croissance des bactéries. Ici, E. coli est incubé à 37oC.
- Après l'incubation, une fois les colonies visibles surgissent, comptez le nombre de colonies bactériennes sur chaque plaque et enregistrez ces valeurs ainsi que leur facteur de dilution associé pour toutes les plaques à chaque point de temps.
- Configuration de l'état de croissance
- Inoculation des médias
3. Analyse des données et résultats
- Densité optique (OD600) Courbe de croissance Terrain
- Tracer la densité optique (OD600) par rapport au temps sur une échelle semi-log. (Figure 3)
- Parcelle de courbe de croissance de l'unité de formation des colonies (CFU/mL)
- Pour chaque point de temps, choisissez la plaque de dilution où le nombre de colonies se situe dans la fourchette de 30 à 300 bactéries. Multipliez le nombre de nombres de colonies par le facteur de dilution, puis par 10, car l'écart de 100 L est considéré comme une dilution supplémentaire de 1:10 lors du calcul de l'UFC/mL.

- Tracer la colonie formant des unités par rapport au temps sur une échelle semi-log. (Figure 4)
- Pour chaque point de temps, choisissez la plaque de dilution où le nombre de colonies se situe dans la fourchette de 30 à 300 bactéries. Multipliez le nombre de nombres de colonies par le facteur de dilution, puis par 10, car l'écart de 100 L est considéré comme une dilution supplémentaire de 1:10 lors du calcul de l'UFC/mL.
- Liaison de la densité optique et des unités de formation de colonies
- Tracer la colonie formant des unités par rapport à la densité optique sur une échelle linéaire pour les lectures OD600 inférieures ou égales à 1,0 OD600 car la relation entre OD600 et CFU/mL est moins précise au-delà de 1,0 OD600. Ici, les six premiers chronopoints sont tracés. (Figure 5)
- Générer une ligne de tendance de régression linéaire affichant l'équation et la valeur R2.
- Déterminer le temps de doublement des bactéries
- À l'aide de la parcelle de courbe de croissance de l'unité de formation de la colonie, pendant la phase exponentielle, identifiez deux points sur le graphique avec la pente la plus raide entre eux pour calculer le temps de doublement.
- Calcul du temps de doublement

- Temps t2 - t1, où t1 - Timepoint 1 et t2 - Timepoint 2
-
 , où b - nombre de bactéries à t2, B - nombre de bactéries à t1, et n - nombre de générations. Dérivé de:
, où b - nombre de bactéries à t2, B - nombre de bactéries à t1, et n - nombre de générations. Dérivé de: .
- Calculez le temps de doublement en utilisant :


 , où b - nombre de bactéries à t2, B - nombre de bactéries à t1, et n - nombre de générations. Dérivé de:
, où b - nombre de bactéries à t2, B - nombre de bactéries à t1, et n - nombre de générations. Dérivé de: Les bactéries se reproduisent par un processus appelé division cellulaire, qui se traduit par deux cellules filles identiques. Si les conditions de croissance sont favorables, les populations bactériennes augmenteront de façon exponentielle.
Les courbes de croissance bactérienne tracent la quantité de bactéries dans une culture en fonction du temps. Une courbe de croissance typique progresse à travers quatre étapes : phase de décalage, phase exponentielle, phase stationnaire et phase de mort. La phase de décalage est le temps qu'il faut aux bactéries pour atteindre un état où elles peuvent se développer et se diviser rapidement. Après cela, la transition des bactéries vers la phase exponentielle, caractérisée par une croissance et une division cellulaires rapides. Le taux de croissance exponentielle de la culture bactérienne au cours de cette phase peut être exprimé comme le temps de doublement, le taux le plus rapide auquel les bactéries peuvent se reproduire dans des conditions spécifiques. La phase stationnaire vient ensuite, où la croissance des cellules bactériennes plateaux et les taux de croissance et de mortalité s'équilibrent en raison de l'épuisement des nutriments environnementaux. Enfin, la bactérie entre dans la phase de la mort. C'est là que la croissance bactérienne diminue fortement et l'épuisement des nutriments graves conduit à la lysing des cellules.
Deux techniques peuvent être utilisées pour quantifier la quantité de bactéries présentes dans une culture et tracer une courbe de croissance. Le premier d'entre eux est par l'intermédiaire d'unités de formation de colonies, ou CUS. Pour obtenir des UFC, une série de neuf dilutions est effectuée à des moments réguliers. La première de ces dilutions, négative dans cet exemple, contient 9mL de PBS et 1mL de la culture bactérienne. Résultat en un facteur de dilution 1:10. Ensuite, 1mL de cette solution est transférée au tube suivant, négatif deux, résultant en un facteur de dilution 1:100. Ce processus se poursuit à travers le dernier tube, né gatif neuf, résultant en un facteur de dilution finale de 1:1 milliard. Après cela, 100 microlitres de chaque dilution sont plaqués. Les plaques sont ensuite incubées et les colonies clonales sont comptées. La plaque de dilution pour un point de temps donné qui pousse entre 30 et 300 colonies est utilisée pour calculer les UFC par millilitre pour ce point de temps.
La deuxième méthode courante de mesure de la concentration bactérienne est la densité optique. La densité optique d'une culture peut être mesurée instantanément, par rapport à un vide média, avec un spectrophotomètre. Typiquement une longueur d'onde de 600 nanomètres, également appelée OD600, est utilisée pour ces mesures, qui augmentent à mesure que la densité cellulaire augmente. Bien que la densité optique soit moins précise que les UFC, elle est pratique car elle peut être obtenue instantanément et nécessite relativement peu de réactifs. Les deux techniques peuvent être utilisées ensemble pour créer une courbe standard qui se rapproche plus précisément du nombre de cellules bactériennes d'une culture. Dans cette vidéo, vous apprendrez comment obtenir des mesures CFUs et OD600 à partir de dilutions en série chronométrées de E. coli. Ensuite, deux courbes de croissance utilisant les mesures CFU et OD600, respectivement, seront tracées avant d'être liées par une courbe standard.
Lorsque vous travaillez avec des bactéries, il est important d'utiliser l'équipement de protection individuelle approprié comme une blouse de laboratoire et des gants et d'observer la technique aseptique appropriée.
Après cela, stériliser le poste de travail avec 70% d'éthanol. Tout d'abord, préparer le bouillon LB et LB solide agar media dans des bouteilles autoclaveables séparées. Après avoir partiellement fermé les bouchons des bouteilles, stériliser les supports dans un autoclave réglé à 121 degrés Celsius pendant 35 minutes. Ensuite, laissez le support d'agar refroidir dans un bain d'eau réglé à 50 degrés Celsius pendant 30 minutes. Une fois refroidi, verser de 20 à 25 ml dans chaque plat Petri. Après cela, laisser les plaques à reposer pendant 24 heures à température ambiante.
Pour préparer les isolats d'une seule colonie qui seront plus tard utilisés pour produire une culture bactérienne liquide, utilisez des stocks préalablement congelés et une technique appropriée de placage de stries pour strier E. coli pour l'isolement sur l'agar LB. Incuber le plat à 37 degrés Celsius pendant la nuit. Après cela, refroidir une boucle d'inoculation stérilisée flamme sur l'agar avant de choisir une seule colonie de la plaque strié. Inoculer 4 ml de support liquide dans un tube à essai de 15 ml. Ensuite, faites pousser la bactérie E. coli à 37 degrés Celsius pendant la nuit en secouant à 210 tr/min.
Pour mettre en place le volume 1:1000 de la culture bactérienne qui sera utilisé dans la courbe de croissance, d'abord obtenir un flacon autoclaved 500 mL Erlenmeyer. Ensuite, utilisez une pipette sérologique de 50 ml pour transférer 100 ml de support stérile sur le flacon. Ensuite, étiquetez neuf tubes à essai de 15 ml consécutivement comme un à neuf. Ces nombres correspondent au facteur de dilution qui sera utilisé pour calculer l'unité de formation de colonie, ou CFU. Ensuite, ajouter 9 ml de 1X PBS à chaque tube. Après cela, étiquetez les plaques d'agar préparées avec les points de temps correspondants et les facteurs de dilution qui seront cultivés. Dans cet exemple avec E. coli, après le point de départ, les points de temps sont pris une fois toutes les heures. En utilisant la culture e. coli liquide liquide préparée au cours de la nuit, inoculer les médias dans le flacon Erlenmeyer autoclave de 500 ml avec un volume de culture de 1:1000. Faites tourbillonner les médias pour répartir uniformément les bactéries.
Après avoir effacé un spectrophotomètre, nettoyez la cuvette avec une lingette sans papier. Ensuite, dispenser 1 ml de la culture dans la cuvette et la placer dans le spectrophotomètre pour obtenir la densité optique de la culture au point zéro. Ensuite, faites pousser la bactérie E. coli à 37 degrés Celsius en secouant à 210 tr/min. À chaque point après le point de temps zéro, retirez un autre 1 ml de culture bactérienne du flacon et répétez la mesure de densité optique. Si la lecture de densité optique est supérieure à 1,0, diluer 100 microlitres de culture bactérienne avec 900 microlitres de médias frais, puis mesurer la densité optique une fois de plus. Cette valeur peut être multipliée par 10 pour la mesure OD 600.
Pour obtenir la mesure de l'unité de formation de la colonie pour chaque point de temps, retirez un ml supplémentaire de culture bactérienne du flacon à chaque moment. Distribuez la culture bactérienne dans le tube à essai négatif et le vortex à mélanger. Ensuite, effectuer la série de dilution en transférant d'abord 1 mL du tube négatif dans les deux tubes négatifs et le vortex pour mélanger. Transférer 1 ml du tube négatif deux dans les trois tubes négatifs et le vortex pour mélanger. Continuer ce transfert en série vers le bas tous les tubes de dilution à la négative neuf tube. Distribuez 100 microlitres de suspension cellulaire sur la plaque étiquetée correspondante pour chaque dilution. Pour chaque dilution, stériliser un épandeur cellulaire dans l'éthanol, le passer à travers une flamme de brûleur Bunsen, et le refroidir en touchant la surface de l'agar loin de l'inoculate. Ensuite, utilisez l'épandeur cellulaire pour étendre la suspension cellulaire jusqu'à ce que la surface de la plaque d'agar devienne sèche. Incuber les plaques à l'envers à 37 degrés Celsius. Une fois que des colonies visibles surgissent, comptez le nombre de colonies bactériennes sur chaque plaque. Enregistrez ces valeurs et leurs facteurs de dilution associés pour chaque plaque à chaque moment.
Pour créer une courbe de croissance OD 600, après s'être assuré que tous les points de données sont entrés correctement dans un tableau, sélectionnez tous les points de temps et leurs données correspondantes. Pour générer une parcelle de courbe de croissance unitaire de formation de colonie, choisissez la plaque de dilution où le nombre de colonies se situe dans la fourchette de 30 à 300 bactéries pour chaque point de temps. Multipliez le nombre de nombres de colonies par le facteur de dilution, puis par dix. C'est parce que la propagation de 100 microlitres est considérée comme une dilution supplémentaire 1:10 lors du calcul des unités de formation de colonie par millilitre. Après cela, tracer la colonie formant des unités par rapport au temps sur une échelle semi-log.
Ces parcelles produites avec des mesures OD 600 et CFU, respectivement, peuvent fournir des informations précieuses sur la cinétique de croissance d'E. coli. La densité optique et les unités de formation de colonie peuvent être liées, de sorte que des CFUs par millilitre peuvent être estimés à partir des mesures oD 600, économisant du temps et des matériaux dans de futures expériences.
Pour ce faire, tracez la colonie formant des unités contre la densité optique sur une échelle linéaire pour les lectures OD 600 inférieures ou égales à 1. 0. Après cela, générer une ligne de tendance de régression linéaire dans le format Y et MX et B, où M est la pente et B est le y-intercept. Cliquez à droite sur les points de données et sélectionnez ajouter la ligne de tendance et linéaire. Ensuite, cochez la case pour afficher l'équation sur le graphique et affichez la valeur au carré R sur le graphique. La valeur au carré R est la mesure statistique de la mesure de la mesure dans quelle mesure les données correspondaient à la ligne de régression ajustée. Dans cet exemple, les 6 premiers points de temps sont tracés avec OD 600 sur l'axe x et CFUs par millilitre sur l'axe y. Dans les expériences futures avec les mêmes conditions de croissance, ces valeurs de pente et y-intercept peuvent être branchées à cette équation pour estimer les UFC à partir des lectures oD 600. Ensuite, regardez la parcelle de courbe de croissance de la colonie formant une unité. Pendant la phase exponentielle, identifiez deux points de temps avec la pente la plus raide entre eux. Pour calculer le temps de doublement, calculez d'abord la variation de temps entre les points de temps sélectionnés. Ensuite, calculez le changement en générations à l'aide de l'équation montrée ici. En l'espèce, le cas le plus faible b est le nombre de bactéries au moment où le troisième point et le cas B supérieur est le nombre de bactéries au moment où le deuxième point. Enfin, divisez le changement dans le temps par le changement en générations. Dans cet exemple, le temps de doublement est de 0. 26 heures ou 15 minutes et 19 secondes. La comparaison des temps de doublement entre différents traitements expérimentaux nous permet d'identifier les meilleures conditions de croissance pour une certaine espèce bactérienne. Par conséquent, le traitement avec le temps de doublement le plus bas sera le plus optimal des conditions examinées.
Subscription Required. Please recommend JoVE to your librarian.
Results
Les parcelles d'unités de formation de colonies et la densité optique sont deux façons de visualiser la cinétique de croissance. En déterminant la relation entre CFU/mL et OD600, la parcelle de densité optique fournit également une estimation de CFU/mL au fil du temps. Les conditions qui entraînent le temps de doublement le plus court sont considérées comme optimales pour la croissance des bactéries données.
Subscription Required. Please recommend JoVE to your librarian.
Applications and Summary
Les courbes de croissance sont précieuses pour comprendre la cinétique de croissance et la physiologie des bactéries. Ils nous permettent de déterminer comment les bactéries réagissent dans des conditions de croissance variables ainsi que de définir les paramètres de croissance optimaux pour une bactérie donnée. Les parcelles de densité de forme de colonies et optiques contiennent toutes deux des informations précieuses illustrant la durée de la phase de décalage, la densité cellulaire maximale atteinte et permettant le calcul du temps de doublement bactérien. Les courbes de croissance permettent également de comparer les différentes bactéries dans les mêmes conditions de croissance. En outre, la densité optique fournit un moyen de standardiser les inoculums initiaux, améliorant la cohérence dans d'autres expériences.
Déterminer quelle approche utiliser lors de la conception d'une expérience de courbe de croissance nécessite une considération. En tant que méthode préférée pour générer des courbes de croissance, les parcelles d'unité de formation de colonies reflètent plus fidèlement les nombres de cellules viables dans la culture des lots. Les parcelles d'unité de formation de colonies permettent également de mesurer la croissance bactérienne dans des conditions qui, autrement, interfèreraient avec une mesure de densité optique. Cependant, il s'agit d'un processus plus long, nécessitant une utilisation intensive de réactifs, et doit être effectué manuellement. Les parcelles de densité optique sont moins précises et ne fournissent qu'une estimation des unités de formation de colonies, ce qui nécessite une courbe standard à générer pour chaque bactérie unique. La densité optique est principalement utilisée pour sa commodité car elle prend beaucoup moins de temps et ne nécessite pas beaucoup de réactifs pour effectuer. Ce qui est le plus intéressant pour la densité optique, c'est que les incubateurs spectrophotométriques peuvent générer automatiquement des courbes de croissance, augmentant considérablement le nombre de conditions de culture qui peuvent être testées à la fois et éliminant la nécessité d'assister constamment à la culture.
Subscription Required. Please recommend JoVE to your librarian.
References
- R. E. Buchanan. 1918. Life Phases in a Bacterial Culture. J Infect Dis 23:109-125.
- CAMPBELL A. 1957. Synchronization of cell division. Bacteriol Rev 21:263-72.
- Wang P, Robert L, Pelletier J, Dang WL, Taddei F, Wright A, Jun S. 2010. Robust growth of Escherichia coli. Curr Biol 20:1099-103.
- Goldman E, Green LH. 2015. Practical Handbook of Microbiology, Third Edition. CRC Press.
- Ben-David A, Davidson CE. 2014. Estimation method for serial dilution experiments. J Microbiol Methods 107:214-221.
- Koch AL. 1968. Theory of the angular dependence of light scattered by bacteria and similar-sized biological objects. J Theor Biol 18:133-156.
- Sezonov G, Joseleau-Petit D, D'Ari R. 2007. Escherichia coli physiology in Luria-Bertani broth. J Bacteriol 189:8746-9.