



Overview
Source: Natalia Martin1, Andrew J. Van Alst1, Rhiannon M. LeVeque1, and Victor J. DiRita1
1 Department of Microbiology and Molecular Genetics, Michigan State University
Bacteria have the ability to exchange genetic material (DeoxyriboNucleic Acid, DNA) in a process known as horizontal gene transfer. Incorporating exogenous DNA provides a mechanism by which bacteria can acquire new genetic traits that allow them to adapt to changing environmental conditions, such as the presence of antibiotics or antibodies (1) or molecules found in natural habitats (2). There are three mechanisms of horizontal gene transfer: transformation, transduction, and conjugation (3). Here we will focus on transformation, the ability of bacteria to take up free DNA from the environment. In the laboratory, the transformation process has four general steps: 1) Preparation of competent cells, 2) Incubation of competent cells with DNA, 3) Recovery of cells, and 4) Plating of the cells for growth of the transformants (Figure 1).
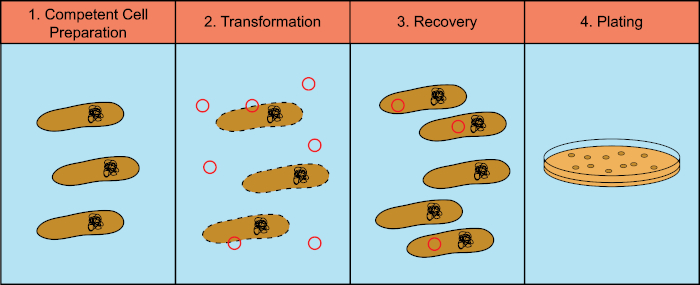
Figure 1: General steps of the transformation process. The transformation process has four general steps: 1) Preparation of competent cells, 2) Incubation with DNA, 3) Recovery of the cells and 4) Plating cells for growth of the transformants.
For transformation to occur, the recipient bacteria must be in a state known as competence. Some bacteria have the ability to become naturally competent in response to certain environmental conditions. However, many other bacteria do not become competent naturally, or the conditions for this process are yet unknown. The ability to introduce DNA into bacteria has a range of research applications: to generate multiple copies of a DNA molecule of interest, to express large amount of proteins, as a component in cloning procedures, and others. Because of the value of transformation to molecular biology, there are several protocols aimed to make cells artificially competent when conditions for natural competence are unknown. Two main methods are used to prepare artificially competent cells: 1) through chemical treatment of cells and 2) exposing the cells to electric pulses (electroporation). The former uses different chemicals depending on the procedure to create attraction between the DNA and the cell surface, while the latter uses electric fields to generate pores in the bacterial cell membrane through which DNA molecules can enter. The most efficient approach for chemical competence is incubation with divalent cations, most notably calcium (Ca2+) (4,5) Calcium-induced competence is the procedure that will be described here (6). This method is mainly used for transformation of Gram-negative bacteria, and that will be the focus of this protocol.
The procedure of chemical transformation involves a series of steps in which cells are exposed to cations to induce chemical competence. These steps are subsequently followed by a temperature change - heat shock - that favors the uptake of foreign DNA by the competent cell (7). The bacterial cell envelopes are negatively charged. In Gram-negative bacteria like Escherichia coli, the outer membrane is negatively-charged due to the presence of lipopolysaccharide (LPS) (8). This results in repulsion of the similarly negatively-charged DNA molecules. In chemical competence induction, positively-charged calcium ions neutralize this charge repulsion enabling DNA absorbance onto the cell surface (9). Calcium treatment and incubation with DNA are done on ice. Subsequently, an incubation at higher temperatures (42°C), the heat shock, is performed. This temperature imbalance further favors the DNA uptake. Bacterial cells need to be at the mid-exponential growth phase to withstand the heat shock treatment; in other growth stages the bacterial cells are too sensitive to the heat resulting in loss of viability which significantly decreases transformation efficiency.
Different DNA sources can be used for transformation. Typically, plasmids, small circular, double-stranded DNA molecules, are used for transformation in most laboratory procedures in E. coli. For plasmids to be maintained in the bacterial cell after transformation, they need to contain an origin of replication. This allows them to be replicated in the bacterial cell independently from the bacterial chromosome. Not all the bacterial cells get transformed during the transformation procedure. Thus, transformation yields a mixture of transformed cells and non-transformed cells. To distinguish between these two populations, a selection method to identify the cells that have acquired the plasmid is used. Plasmids usually contain selectable markers, which are genes encoding a trait that confers an advantage for growth (i.e. resistance to an antibiotic or chemical or rescue from a growth auxotrophy). After transformation, bacterial cells are plated on selective media, which only allows for growth of the transformed cells. In the case of cells transformed with a plasmid conferring resistance to a given antibiotic, the selective media will be growth media containing that antibiotic. Several different methods can be used to confirm that the colonies grown in the selective media are transformants (i.e. have incorporated the plasmid). For example, plasmids can be recovered from these cells using plasmid preparation methods (10) and digested to confirm plasmid size. Alternatively, colony PCR can be used to confirm the presence of the plasmid of interest (11).
The aim of this experiment is to prepare E. coli DH5α chemically competent cells, using an adaptation of the calcium chloride procedure (12), and to transform them with the plasmid pUC19 to determine transformation efficiency. The E. coli strain DH5α is a commonly used strain in molecular biology applications. Because of its genotype, specifically the recA1 and endA1, this strain allows for increased insert stability and improve the quality of plasmid DNA in subsequent preparations. Since the transformation efficiency decreases with increasing size of the DNA, the plasmid pUC19 was used in this protocol because of its small size (2686 bp) (see https://www.mobitec.com/cms/products/bio/04_vector_sys/standard_cloning_vectors.html for a vector map). pUC19 confers resistance to ampicillin and thus, this was the antibiotic used for selection.
Procedure
This protocol describes the preparation and transformation of competent E. coli DH5α using an adaptation of the calcium chloride procedure (12).
1. Set-up
- Equipment
- Spectrophotometer
- Sorval Centrifuge (or equivalent)
- Benchtop centrifuge
- Heat block or water bath
- Orbital Shaker
- Stationary Incubator
- Gel casting tray
- Well combs
- Voltage source
- Gel box
- UV light source
- Microwave
- Solutions and reagents
- Luria-Bertani (LB) broth (10 g casein enzymic hydrolysate, 5 g yeast extract and 5 g sodium chloride in 1000 mL of H2O)
- Super Optimal broth with Catabolite repression (SOC): (2% (w/v) tryptone, 0.5% (w/v) yeast extract, 10 mM NaCl, 2.5 mM KCl, 10 mM MgCl2, 10 mM MgSO4, and 20 mM glucose)
- CaCl2-MgCl2 (80 mM MgCl2, 20 mM CaCl2) solution.
- M CaCl2 solution (if cells will be transformed immediately) or 0.1M CaCl2 solution containing 10% (v/v) glycerol (if cells will be frozen for future use).
- LB agar plates
- LB agar selective plates (for this experiment, since the plasmid used confers ampicillin resistance, LB agar plates containing ampicillin 100 µg/mL were used)
- E. coli DH5α strain
- Plasmid pUC19 DNA (100 pg/ µl)
- QIAprep Spin Miniprep Kit (Qiagen)
- HindIII restriction enzyme
- 1 kb plus DNA ladder
- Low Melting Point Agarose
- 1X TAE buffer (40 mM Tris Base, 20 mM Acetic Acid and 1mM EDTA)
- Ethidium Bromide (10mg/mL)
- General safety notes
E. coli DH5α is classified as Biosafety Level 1 (BSL1). Microbes in this category pose little to no threat of infection in healthy adults. However, careful manipulation of the microorganism is required.
IMPORTANT all steps in this protocol need to be carried out using aseptic techniques and on ice or 4°C temperatures unless indicated.
2. Protocol
- From a frozen stock of E. coli DH5α (frozen in 20% glycerol from an overnight culture grown in LB) streak out bacteria for isolation on an LB agar plate. Incubate at 37°C overnight (16-20 hours).
- Inoculate a single colony into 3 mL of LB broth in a tube. Grow shaking at 210 rpm at 37°C overnight (16-20 hours).
- Measure the OD600 of the overnight culture. Use the overnight culture to inoculate 100 mL of LB broth in a 1-liter flask to an OD600=0.01. Incubate the culture shaking vigorously (210 rpm) at 37°C monitoring OD600 in the spectrophotometer every 15-20 min, until culture reaches OD600=0.35 (approximately 3 hours).
NOTE: For transformation to be efficient, bacterial cells need to be at mid-exponential growth phase. The maximum number of cells needs to be 108 cells/mL, which for most strains of E. coli corresponds to OD600=0.4. The use of the spectrophotometer allows to measure the OD600, which allows to determine that the cells are at the appropriate growth stage. If this protocol will be used for other strains of bacteria, calibration to determine the number of colonies forming units at specific OD600 values will be necessary to determine this correlation. - Transfer the 50 mL of the culture to each of 2 ice-cold polypropylene centrifuge bottles. Place the bottles on ice for 20 min to cool.
- Recover the cells by centrifugation at 2700g (4100 rpm in a Sorval GSA rotor) for 10 min at 4°C.
- Remove the supernatant. Drain away the last traces of media by placing the bottle upside down on a pad or paper towel.
- Resuspend each bacterial pellet into 30 mL of a CaCl2-MgCl2 (80 mM MgCl2, 20 mM CaCl2) ice-cold solution. First add 5 mL of the solution, swirl carefully until pellet has dissolved completely and then add the remaining 25 mL of solution.
- Repeat step 2.4.
- Repeat step 2.5.
- If competent cells are going to be directly transformed, resuspend each bacterial pellet into 2 mL of a CaCl2 (0.1 M) ice-cold solution by swirling the tubes carefully. If the pellet does not get resuspended with this method, resuspend by gently pipetting up and down (avoiding bubble formation).
Alternatively, the competent cells can be frozen and stored for later use. To prepare frozen stocks of competent cells, resuspend the pellet in 2 ml of a 0.1M CaCl2 solution containing 10% (v/v) glycerol. This solution needs to be ice-cold. Aliquot cell suspension into ice-cold 1.5 mL polypropylene tubes (160 µl per tube). Freeze competent cells immediately in a dry ice/ethanol bath. Transfer tubes to a -70°C freezer. - To transform the CaCl2-treated cells, transfer 50 µl of competent cells to each of 2 1.5 ml polypropylene tubes. Add the 1 µl (100 pg) of pUC19 plasmid DNA to one of the tubes and leave the second tube without DNA (negative control). Mix gently (avoid bubble formation). Incubate for 30 min on ice.
NOTE: No more than 50 ng of DNA in a volume of 10 µL or less should be used in the transformation. - Transfer the tubes to the heat block and incubate at 42°C for 45 s exactly.
NOTE: Heat shock is a critical step. Do not exceed temperature or incubation time. - Readily transfer tubes to ice. Incubate for 2 min.
- Add 950 µL of SOC media and incubate the tubes for 1 hour at 37°C to allow the bacteria to recover and express the antibiotic resistant marker encoded in the plasmid.
- Dilute 10 µL of the cell suspension in 1000 µL in SOC (1/100 dilution) and 100 µL of the cell suspension in 1000 µL in SOC (1/10 dilution). Plate 100 µl of the dilutions, as well as the control, onto selective plates, and spread using a spatula. Usually, plating 100 µL of a 1/100 and 1/10 dilution will yield enough number of colony forming units (cfu) per plate. Ideally, this number should range between 30-300 cfu so that there are enough colonies but separated from each other. However, the number of cfu will depend on the transformation efficiency (see Data Analysis and Results Section).
- Incubate the plates at 37°C. Transformed colonies should appear in 12-16 hours (this range will depend on the cell strain and selection method). No colonies should grow in the negative control.
- Count the cfu/plate obtained for the transformation (Table 1).
- To verify the transformants harbor the pUC19 plasmid, a plasmid preparation and subsequent digestion will be performed. To this end, inoculate a single colony into 3 ml of LB broth in a tube. Grow shaking at 210 rpm at 37°C overnight (16-20 hours).
- Prepare a plasmid preparation using the QIAprep Spin Miniprep Kit, according to the instructions from the manufacturer.
- Digest the 1 µg of purified pUC19 with the restriction enzyme HindIII at 37°C for 1 hour.
NOTE: Any enzyme that cuts in the pUC19 multiple cloning site can be used for this step.
| Component | Amount |
| 10X Restriction digest buffer | 2.5 µl |
| Plasmid pUC19 | 1 µg |
| HindIII | 1 µl |
| H2O | 20.5 µl (to 25 µl) |
- Run a molecular weight ladder, digested pUC19 DNA and the same amount of undigested pUC19 DNA in a 1% agarose gel containing 1 µg/mL ethidium bromide for 1 hour at 95 V.
NOTE: time and voltage will vary depending on equipment used. - Visualize gel under UV light. Compare size of digested and undigested pUC19 DNA (Figure 2) (see Data Analysis and Results Section).
Proceed with the necessary steps required to verify the transformation according to the goal of each particular transformation experiment.

Figure 2: Digestion of recovered plasmid DNA from transformed DH5α cells. Plasmid DNA was recovered from transformed DH5α cells, digested with HindIII, run in a 1% agarose gel and visualized with a UV source (steps 2.19 to 2.22).
3. Data Analysis and Results
To calculate the transformation efficiency, an indicator of how well the cells took up the extracellular DNA, the colonies obtained in the transformation need to be counted:
| Dilution | Cfu |
| 1/100 | 34 |
| 1/10 | 246 |
Table 1: Colony forming units (cfu) counted from transformation experiment.
Transformation efficiency (TE) is a measure of the number of cfu resulting from transforming 1 µg of plasmid into a given volume of competent cells. Many parameters affect the transformation efficiency: plasmid size, cell genotype, growth stage during competence preparation, methods of transformation, etc.). When calculating the TE is important to consider what dilution (if any) was performed before plating and incorporate it in the calculation of the total number of cfu. The transformation efficiency (TE) is calculated with the following equation:

First divide the cfu by the µg of DNA, in this example 0.0001µg. Then divide the result by the dilution factor. In this example, a 1/10 dilution was used and 100µL of a 1 ml solution was plated (dilution: 1/10 × 100 µL /1000 µL=0.01).

Bacteria are remarkably adaptable and one mechanism which facilitates this adaptation is their ability to take in external DNA molecules. One type of DNA that bacteria can uptake is called a plasmid, a circular piece of DNA that frequently contains useful information, such as antibiotic resistance genes. The process of bacteria being modified by new genetic information incorporated from an external source is referred to as transformation. Transformation can easily be performed in the laboratory using Escherichia coli, or E. coli.
In order to be transformed, E. coli cells must first be made competent, which means capable of taking in DNA molecules from their environment. The protocol for accomplishing this is surprisingly simple, a short incubation of the cells in a calcium chloride solution. This incubation causes the cells to become permeable to DNA molecules. After the cells are pelleted by centrifugation, the supernatant is removed. The plasmid DNA is now added to the competent cells. After incubating the cells with DNA, the mix is briefly heated to 42 degrees Celsius, followed by rapid cooling on ice. This heat shock causes the DNA to be transferred across the cell's wall and membranes. The cells are then incubated in fresh media. Then, the bacteria are placed at 37 degrees to allow them to reseal their membranes and express resistant proteins.
Those cells which have taken in the plasmids will faithfully copy the DNA and pass it to their progeny and express any proteins that might be encoded by it, including antibiotic resistance mediators. Those resistance genes can be used as selectable markers to identify bacteria which have been successfully transformed because cells that have not taken up the plasmid will not express the resistance gene product. This means that when the cells are plated on a solid medium which contains the appropriate antibiotic, only cells that have taken up the plasmid will grow. Transformation of the cells in a growing colony can be further confirmed by culturing those cells in liquid media overnight to increase the yield before extracting the DNA from the sample. Once the DNA is isolated, a diagnostic restriction enzyme digest can be carried out. Because restriction enzymes cut DNA in predictable locations, running these digests on a gel should show a predictable pattern if the desired plasmid was successfully transformed. For example, if pUC19 is prepared and cut with the restriction enzyme HindIII, a single band of 2686 nucleotides should be seen on the gel.
In this lab, you will transform E. coli strain DH-5 Alpha with pUC19, and then confirm the successful transformation by DNA gel electrophoresis.
Before starting the procedure, put on the appropriate personal protective equipment, including a lab coat and gloves. Next, sterilize the workspace with 70% ethanol.
Now, prepare chemically competent cells by depositing a loopfull of bacteria onto a sterile LB agar plate and streaking the bacteria with a new loop. Then, incubate the plate at 37 degrees Celsius overnight. The next day, sterilize the bench top with 70% ethanol again, and remove the plate from the incubator.
Inoculate a single, well-isolated colony into 3 milliliters of LB broth in a tube with a sterile loop. Then, grow the culture at 37 degrees Celsius overnight, with shaking at 210 RPM. The next day, measure the optical density of the overnight culture with a spectrophotometer. Then, add 100 milliliters of LB broth to a one-liter flask, and inoculate it with the overnight culture at an optical density of 0. 01. Now, incubate the culture at 37 degrees Celsius with shaking, and check the OD600 every 15 to 20 minutes until the culture reaches mid-exponential growth phase.
After approximately three hours, transfer 50 milliliters of the culture to two ice-cold polypropylene bottles. Then, place the bottles back on ice for 20 minutes to cool. Next, recover the cells via centrifugation. Discard the supernatants and place the bottles upside down on a paper towel. Next, resuspend the bacterial pellet in five milliliters of ice-cold calcium chloride magnesium chloride solution and swirl carefully until the pellet has dissolved completely. Then, add another 25 milliliters of the solution to the dissolved bacterial pellet. Resuspend the other bacterial pellet as previously demonstrated. After this, repeat the centrifugation, and remove the supernatants.
If the competent cells are going to be directly transformed, resuspend each bacterial pellet in two milliliters of an ice-cold 0.1 molar calcium chloride solution by swirling the tubes carefully. To begin the transformation procedure, transfer 50 microliters of competent cells to two labeled 1.5 milliliter polypropylene tubes. Then, add one microliter of pUC19 plasmid DNA to one of the tubes. Mix gently, avoiding bubble formation, and incubate both tubes for 30 minutes on ice. After incubation, transfer the tubes to a heat block and incubate at 42 degrees Celsius for 45 seconds. Immediately transfer the tubes to ice, and incubate for two minutes. Now, add 950 microliters of SOC media to each tube and incubate them for one hour at 37 degrees Celsius to allow the bacteria to recover, and express the antibiotic resistant marker encoded in the plasmid.
To make a 1 to 100 dilution, add 990 microliters of SOC media and 10 microliters of cell suspension to a 1.5 milliliter tube. Then, make a 1 to 10 dilution by adding 900 microliters of SOC media and 100 microliters of cell suspension to a 1.5 milliliter tube. Next, plate 100 microliters of the diluted cell suspensions and 100 microliters of the negative control, onto separate selective plates containing ampicillin using a spreader and incubate the plates at 37 degrees Celsius for 12 to 16 hours. After incubation, count the colony-forming units, or CFUs, per plate, obtained through transformation, and record these data. To verify that the transformants have the pUC19 plasmid, pick a single, well-isolated colony from a plate with a sterile loop, and introduce it to a tube containing 3 milliliters of LB broth. Then, incubate the culture at 37 degrees Celsius with shaking, overnight. The next day, use a DNA mini prep kit to isolate DNA from 3 milliliters of the culture, according to the manufacturer's instructions. After completing the DNA mini prep, digest the 1 microgram of purified pUC19 with a restriction enzyme at 37 degrees Celsius for 1 hour. Now, load 20 microliters of a molecular weight ladder, 1 microgram of digested plasmid DNA, and 1 microgram of undigested plasmid DNA into consecutive wells of a 1% agarose gel containing 1 microgram per milliliter ethidium bromide. Then, run the gel for 1 hour at 95 volts. Finally, visualize the gel with a UV illuminator.
In this experiment, E. coli DH5 Alpha chemically competent cells were prepared using an adaptation of the calcium chloride procedure, and then transformed with the plasmid pUC19 to determine transformation efficiency. To calculate the transformation efficiency, use the recorded CFU counts for the 1 in 100 and 1 in 10 dilutions, and any other dilutions with CFU counts between 30 and 300. First, the recorded CFU count, 246 in this example, is divided by the amount of DNA, .0001 micrograms here, that was plated. Then, this number is divided by the dilution factor used to give the transformation efficiency in CFUs per microgram. In this example, a 1 to 10 dilution was used and 100 microliters of a 1 milliliter solution was plated, giving a final dilution factor of 0.01. In the undigested plasmid lane, the circular DNA may appear as two or three different bands of varying brightness. This is because the circular, uncut DNA may exist in several different conformation states, such as supercoiled, open circle, or more linear, and each of these move through the gel at different rates. Analysis of the recovered plasmid DNA digestion indicated that the plasmid used has an expected size of pUC19 DNA, 2,686 base pairs.
Subscription Required. Please recommend JoVE to your librarian.
Results
Although TE is dependent on many factors, non-commercial competent cell preparations, like this one, normally yield 106 to 107 transformants per microgram of plasmid. Therefore, this preparation, with a TE = 2.46 x 108 cfu/µg, yielded a TE well beyond the expected range. Additional protocols are available for making supercompetent cells when higher transformation efficiencies are required for a given application (13).
Analysis of the digestion of the plasmid DNA recovered from the transformed cells indicated that this plasmid has the expected size of pUC19 DNA (2686 bp).
Subscription Required. Please recommend JoVE to your librarian.
Applications and Summary
Transformation is a powerful method for introducing exogenous DNA into bacterial cells that is key to many molecular biology applications in the laboratory. Additionally, it plays a major role in nature by allowing bacterial cells to exchange genetic material that could result in increased genetic variation and allow for acquisition of different beneficial traits for survival under a wide range of conditions. Many bacterial strains encode the genes required for natural competence. However, the conditions in which these genes are induced are still unknown. Further research is required to determine these conditions.
Subscription Required. Please recommend JoVE to your librarian.
References
- Croucher, N. J. et al. Rapid pneumococcal evolution in response to clinical interventions. Science. 331 (6016):430-434. (2011)
- Borgeaud, S. et al. The type VI secretion system of Vibrio cholerae fosters horizontal gene transfer. Science. 347(6217):63-67. (2015)
- Burmeister, A. R. Horizontal Gene Transfer. Evol Med Public Health. 2015 (1):193-194. (2015)
- Weston A, Brown MG, Perkins HR, Saunders JR, Humphreys GO. Transformation of Escherichia coli with plasmid deoxyribonucleic acid: calcium-induced binding of deoxyribonucleic acid to whole cells and to isolated membrane fractions. J Bacteriol. 145 (2):780-7. (1981)
- Dagert M, Ehrlich SD. Prolonged incubation in calcium chloride improves the competence of Escherichia coli cells. Gene. 6 (1):23-8. (1979)
- Asif A, Mohsin H, Tanvir R, and Rehman Y. Revisiting the Mechanisms Involved in Calcium Chloride Induced Bacterial Transformation. Front Microbiol. 8:2169. (2017)
- Panja S, Aich P, Jana B, Basu T. How does plasmid DNA penetrate cell membranes in artificial transformation process of Escherichia coli? Mol Membr Biol. 25 (5):411-22. (2008)
- Silhavy, TJ, Kahne D, Walker S. The Bacterial Cell Envelope. Cold Spring Harb Perspect Biol. 2 (5): a000414. (2010)
- Panja S, Aich P, Jana B, Basu T. (2008) Plasmid DNA binds to the core oligosaccharide domain of LPS molecules of E. coli cell surface in the CaCl2-mediated transformation process. Biomacromolecules. 9 (9):2501-9.
- JoVE Science Education Database. Basic Methods in Cellular and Molecular Biology. Plasmid Purification. JoVE, Cambridge, MA. (2018)
- Bergkessel M and Guthrie C. Colony PCR. Methods in Enzymology. 529: 299-309. (2013)
- Sambrook J and Russell DW. Molecular Cloning A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.Protocol 25 (1.116-118). (2001)
- Wirth R, Friesenegger A, Fiedler S. Transformation of various species of gram-negative bacteria belonging to 11 different genera by electroporation. Molecular & General Genetics. 216 (1): 175-7. (1989)