



Overview
Fonte: Natalia Martin1, Andrew J. Van Alst1, Rhiannon M. LeVeque1e Victor J. DiRita1
1 Dipartimento di Microbiologia e Genetica Molecolare, Michigan State University
I batteri hanno la capacità di scambiare materiale genetico (acido desossiribonucleico, DNA) in un processo noto come trasferimento genico orizzontale. L'incorporazione di DNA esogeno fornisce un meccanismo attraverso il quale i batteri possono acquisire nuovi tratti genetici che consentono loro di adattarsi alle mutevoli condizioni ambientali, come la presenza di antibiotici o anticorpi (1) o molecole presenti negli habitat naturali (2). Esistono tre meccanismi di trasferimento genico orizzontale: trasformazione, trasduzione e coniugazione (3). Qui ci concentreremo sulla trasformazione, la capacità dei batteri di prendere il DNA libero dall'ambiente. In laboratorio, il processo di trasformazione ha quattro fasi generali: 1) Preparazione di cellule competenti, 2) Incubazione di cellule competenti con DNA, 3) Recupero di cellule e 4) Placcatura delle cellule per la crescita dei trasformanti (Figura 1).
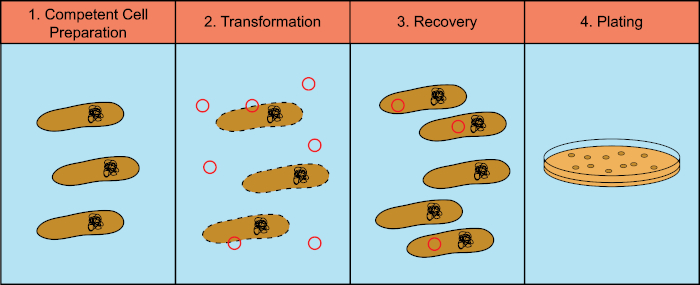
Figura 1: Fasi generali del processo di trasformazione. Il processo di trasformazione ha quattro fasi generali: 1) Preparazione delle cellule competenti, 2) Incubazione con DNA, 3) Recupero delle cellule e 4) Cellule placcate per la crescita dei trasformanti.
Affinché si verifichi la trasformazione, i batteri riceventi devono essere in uno stato noto come competenza. Alcuni batteri hanno la capacità di diventare naturalmente competenti in risposta a determinate condizioni ambientali. Tuttavia, molti altri batteri non diventano competenti naturalmente, o le condizioni per questo processo sono ancora sconosciute. La capacità di introdurre il DNA nei batteri ha una serie di applicazioni di ricerca: generare più copie di una molecola di DNA di interesse, esprimere grandi quantità di proteine, come componente nelle procedure di clonazione e altri. A causa del valore della trasformazione in biologia molecolare, ci sono diversi protocolli volti a rendere le cellule artificialmente competenti quando le condizioni per la competenza naturale sono sconosciute. Due metodi principali sono utilizzati per preparare cellule artificialmente competenti: 1) attraverso il trattamento chimico delle cellule e 2) esponendo le cellule a impulsi elettrici (elettroporazione). Il primo utilizza diverse sostanze chimiche a seconda della procedura per creare attrazione tra il DNA e la superficie cellulare, mentre il secondo utilizza campi elettrici per generare pori nella membrana cellulare batterica attraverso i quali le molecole di DNA possono entrare. L'approccio più efficiente per la competenza chimica è l'incubazione con cationi bivalenti, in particolare calcio (Ca2+)(4,5) La competenza indotta dal calcio è la procedura che verrà descritta qui (6). Questo metodo viene utilizzato principalmente per la trasformazione dei batteri Gram-negativi e questo sarà il focus di questo protocollo.
La procedura di trasformazione chimica comporta una serie di passaggi in cui le cellule sono esposte a cationi per indurre competenza chimica. Questi passaggi sono successivamente seguiti da un cambiamento di temperatura - shock termico - che favorisce l'assorbimento di DNA estraneo da parte della cellula competente (7). Gli involucri cellulari batterici sono caricati negativamente. Nei batteri Gram-negativi come l'Escherichia coli,la membrana esterna è caricata negativamente a causa della presenza di lipopolisaccaride (LPS) (8). Ciò si traduce in repulsione delle molecole di DNA caricate negativamente in modo simile. Nell'induzione della competenza chimica, gli ioni calcio caricati positivamente neutralizzano questa repulsione di carica consentendo l'assorbanza del DNA sulla superficie cellulare (9). Il trattamento del calcio e l'incubazione con dna vengono effettuati su ghiaccio. Successivamente, viene eseguita un'incubazione a temperature più elevate (42°C), lo shock termico. Questo squilibrio di temperatura favorisce ulteriormente l'assorbimento del DNA. Le cellule batteriche devono essere nella fase di crescita media-esponenziale per resistere al trattamento di shock termico; in altre fasi di crescita le cellule batteriche sono troppo sensibili al calore con conseguente perdita di vitalità che diminuisce significativamente l'efficienza di trasformazione.
Diverse fonti di DNA possono essere utilizzate per la trasformazione. Tipicamente, i plasmidi, piccole molecole di DNA circolari a doppio filamento, vengono utilizzati per la trasformazione nella maggior parte delle procedure di laboratorio in E. coli. Affinché i plasmidi possano essere mantenuti nella cellula batterica dopo la trasformazione, devono contenere un'origine di replicazione. Ciò consente loro di essere replicati nella cellula batterica indipendentemente dal cromosoma batterico. Non tutte le cellule batteriche vengono trasformate durante la procedura di trasformazione. Pertanto, la trasformazione produce una miscela di cellule trasformate e cellule non trasformate. Per distinguere tra queste due popolazioni, viene utilizzato un metodo di selezione per identificare le cellule che hanno acquisito il plasmide. I plasmidi di solito contengono marcatori selezionabili, che sono geni che codificano un tratto che conferisce un vantaggio per la crescita (cioè resistenza a un antibiotico o a una sostanza chimica o salvataggio da un'auxotrofia di crescita). Dopo la trasformazione, le cellule batteriche vengono placcate su mezzi selettivi, che consentono solo la crescita delle cellule trasformate. Nel caso di cellule trasformate con un plasmide che conferisce resistenza a un determinato antibiotico, il mezzo selettivo sarà un mezzo di crescita contenente quell'antibiotico. Diversi metodi possono essere utilizzati per confermare che le colonie coltivate nei mezzi selettivi sono trasformanti (cioè hanno incorporato il plasmide). Ad esempio, i plasmidi possono essere recuperati da queste cellule utilizzando metodi di preparazione del plasmide (10) e digeriti per confermare le dimensioni del plasmide. In alternativa, la PCR della colonia può essere utilizzata per confermare la presenza del plasmide di interesse (11).
Lo scopo di questo esperimento è quello di preparare cellule chimicamente competenti di E. coli DH5α, utilizzando un adattamento della procedura del cloruro di calcio (12), e di trasformarle con il plasmide pUC19 per determinare l'efficienza di trasformazione. Il ceppo di E. coli DH5α è un ceppo comunemente usato nelle applicazioni di biologia molecolare. A causa del suo genotipo, in particolare recA1 e endA1, questo ceppo consente una maggiore stabilità dell'inserto e migliora la qualità del DNA plasmidico nei preparati successivi. Poiché l'efficienza di trasformazione diminuisce con l'aumentare delle dimensioni del DNA, il plasmide pUC19 è stato utilizzato in questo protocollo a causa delle sue piccole dimensioni (2686 bp) (vedi https://www.mobitec.com/cms/products/bio/04_vector_sys/standard_cloning_vectors.html per una mappa vettoriale). pUC19 conferisce resistenza all'ampicillina e, quindi, questo era l'antibiotico utilizzato per la selezione.
Procedure
Questo protocollo descrive la preparazione e la trasformazione del competente E. coli DH5α utilizzando un adattamento della procedura del cloruro di calcio (12).
1. Configurazione
-
Attrezzatura
- Spettrofotometro
- Centrifuga Sorval (o equivalente)
- Centrifuga da banco
- Blocco termico o bagno d'acqua
- Agitatore orbitale
- Incubatore stazionario
- Vassoio per colata in gel
- Pettini di pozzo
- Sorgente di tensione
- Scatola di gel
- Sorgente di luce UV
- Microonde
-
Soluzioni e reagenti
- Brodo di Luria-Bertani (LB) (10 g di idrolizzato enzimatico di caseina, 5 g di estratto di lievito e 5 g di cloruro di sodio in 1000 ml di H2O)
- Brodo super ottimale con repressione della catabolite (SOC): (2% (p/v) triptone, 0,5% (p/v) estratto di lievito, 10 mM NaCl, 2,5 mM KCl, 10 mM MgCl2,10 mM MgSO4e 20 mM glucosio)
- CaCl2-MgCl2 (80 mM MgCl2, 20 mM CaCl2) soluzione.
- MCaCl 2 soluzione (se le cellule saranno trasformate immediatamente) o 0,1M soluzione di CaCl2 contenente il 10% (v/v) di glicerolo (se le cellule saranno congelate per un uso futuro).
- Piastre di agar LB
- Piastre selettive LB agar (per questo esperimento, poiché il plasmide utilizzato conferisce resistenza all'ampicillina, sono state utilizzate piastre lb agar contenenti ampicillina 100 μg/mL)
- E. coli Ceppo DH5α
- Plasmide pUC19 DNA (100 pg/ μl)
- Kit QIAprep Spin Miniprep (Qiagen)
- Posteriore III enzima di restrizione
- 1 kb più scala DNA
- Agarose a basso punto di fusione
- 1X tampone TAE (40 mM Tris Base, 20 mM Acido Acetico e 1mM EDTA)
- Bromuro di etidio (10mg/mL)
-
Note generali sulla sicurezza
E. coli DH5α è classificato come Livello di Biosicurezza 1 (BSL1). I microbi in questa categoria rappresentano poca o nessuna minaccia di infezione negli adulti sani. Tuttavia, è necessaria un'attenta manipolazione del microrganismo.
IMPORTANTE tutte le fasi di questo protocollo devono essere eseguite utilizzando tecniche asettiche e su ghiaccio o temperature di 4° C se non indicato.
2. Protocollo
- Da uno stock congelato di E. coli DH5α (congelato al 20% in glicerolo da una coltura notturna coltivata in LB) strisciare i batteri per l'isolamento su una piastra di agar LB. Incubare a 37°C durante la notte (16-20 ore).
- Inoculare una singola colonia in 3 ml di brodo LB in un tubo. Crescere scuotendo a 210 giri/min a 37°C durante la notte (16-20 ore).
- Misurare l'OD600 della cultura notturna. Utilizzare la coltura notturna per inoculare 100 ml di brodo LB in un pallone da 1 litro a un OD600 = 0,01. Incubare la coltura scuotendo vigorosamente (210 rpm) a 37°C monitorando OD600 nello spettrofotometro ogni 15-20 min, fino a raggiungere OD600=0,35 (circa 3 ore).
NOTA: affinché la trasformazione sia efficiente, le cellule batteriche devono essere in fase di crescita media-esponenziale. Il numero massimo di cellule deve essere di 108 cellule/mL, che per la maggior parte dei ceppi di E. coli corrisponde a OD600=0,4. L'uso dello spettrofotometro consente di misurare l'OD600, che consente di determinare che le cellule sono nella fase di crescita appropriata. Se questo protocollo verrà utilizzato per altri ceppi di batteri, sarà necessaria la calibrazione per determinare il numero di colonie che formano unità a valori specifici di OD600 per determinare questa correlazione. - Trasferire i 50 ml della coltura in ciascuna delle 2 bottiglie di centrifuga in polipropilene ghiacciato. Mettere le bottiglie sul ghiaccio per 20 minuti per raffreddare.
- Recuperare le celle mediante centrifugazione a 2700g (4100 rpm in un rotore Sorval GSA) per 10 min a 4°C.
- Rimuovere il surnatante. Scolare via le ultime tracce di supporto posizionando la bottiglia a testa in giù su un tampone o un asciugamano di carta.
- Riconsoppiare ogni pellet batterico in 30 mL di unasoluzioneghiacciata CaCl2 -MgCl2 (80mM MgCl 2 ,20mM CaCl 2 ). Per prima cosa aggiungere 5 ml di soluzione, ruotare con attenzione fino a quando il pellet si è sciolto completamente e quindi aggiungere i restanti 25 mL di soluzione.
- Ripetere il passaggio 2.4.
- Ripetere il passaggio 2.5.
- Se le cellule competenti stanno per essere trasformate direttamente, riconsemere ogni pellet batterico in 2 ml di una soluzione ghiacciata di CaCl2 (0,1 M) facendo roteare attentamente i tubi. Se il pellet non viene riconsociato con questo metodo, risusepenare delicatamente su e giù (evitando la formazione di bolle).
In alternativa, le cellule competenti possono essere congelate e conservate per un uso successivo. Per preparare le scorte congelate di cellule competenti, risuscimere il pellet in 2 ml di una soluzione di 0,1M CaCl2 contenente il 10% (v/v) di glicerolo. Questa soluzione deve essere ghiacciata. Sospensione di cellule aliquote in tubi di polipropilene da 1,5 mL ghiacciati (160 μl per tubo). Congelare immediatamente le cellule competenti in un bagno di ghiaccio secco/etanolo. Trasferire i tubi in un congelatore a -70°C. - Per trasformare le cellule trattate con CaCl2,trasferire 50 μl di cellule competenti in ciascuna delle 2 provette di polipropilene da 1,5 ml. Aggiungere 1 μl (100 pg) di DNA plasmidico pUC19 a uno dei tubi e lasciare il secondo tubo senza DNA (controllo negativo). Mescolare delicatamente (evitare la formazione di bolle). Incubare per 30 minuti sul ghiaccio.
NOTA: Nella trasformazione non devono essere utilizzati più di 50 ng di DNA in un volume pari o inferiore a 10 μL. - Trasferire i tubi nel blocco di calore e incubare a 42°C per 45 s esatti.
NOTA: lo shock termico è un passaggio critico. Non superare la temperatura o il tempo di incubazione. - Trasferire facilmente i tubi sul ghiaccio. Incubare per 2 min.
- Aggiungere 950 μL di mezzi SOC e incubare le provette per 1 ora a 37°C per consentire ai batteri di recuperare ed esprimere il marcatore resistente agli antibiotici codificato nel plasmide.
- Diluire 10 μL della sospensione cellulare in 1000 μL in SOC (diluizione 1/100) e 100 μL della sospensione cellulare in 1000 μL in SOC (diluizione 1/10). Piastra 100 μl delle diluizioni, così come il controllo, su piastre selettive, e diffondere utilizzando una spatola. Di solito, la placcatura di 100 μL di una diluizione 1/100 e 1/10 produrrà un numero sufficiente di unità formanti colonie (cfu) per piastra. Idealmente, questo numero dovrebbe variare tra 30-300 ufc in modo che ci siano abbastanza colonie ma separate l'una dall'altra. Tuttavia, il numero di cfu dipenderà dall'efficienza della trasformazione (vedere la sezione Analisi dei dati e risultati).
- Incubare le piastre a 37°C. Le colonie trasformate dovrebbero apparire in 12-16 ore (questo intervallo dipenderà dal ceppo cellulare e dal metodo di selezione). Nessuna colonia dovrebbe crescere nel controllo negativo.
- Contare il cfu/piastra ottenuto per la trasformazione (Tabella 1).
- Per verificare che i trasformanti ospitino il plasmide pUC19, verrà eseguita una preparazione del plasmide e la successiva digestione. A tal fine, inoculare una singola colonia in 3 ml di brodo LB in un tubo. Crescere scuotendo a 210 giri/min a 37°C durante la notte (16-20 ore).
- Preparare una preparazione del plasmide utilizzando il kit QIAprep Spin Miniprep, secondo le istruzioni del produttore.
- Digerire 1 μg di pUC19 purificato con l'enzima di restrizione HindIII a 37°C per 1 ora.
NOTA: per questo passaggio è possibile utilizzare qualsiasi enzima che taglia nel sito di clonazione multipla pUC19.
| Componente | Importo |
| Buffer digest di restrizione 10X | 2,5 μl |
| Plasmide pUC19 | 1 μg |
| Posteriore III | 1 μl |
| H2O | Da 20,5 μl (a 25 μl) |
- Eseguire una scala a peso molecolare, DIGERIRE IL DNA pUC19 e la stessa quantità di DNA pUC19 non digerito in un gel di agarosio all'1% contenente 1 μg/mL di bromuro di etidio per 1 ora a 95 V.
NOTA: il tempo e la tensione variano a seconda dell'apparecchiatura utilizzata. - Visualizza il gel sotto la luce UV. Confrontare le dimensioni del DNA pUC19 digerito e non digerito (Figura 2) (vedere la sezione Analisi dei dati e risultati).
Procedere con i passaggi necessari per verificare la trasformazione in base all'obiettivo di ogni particolare esperimento di trasformazione.

Figura 2: Digestione del DNA plasmidico recuperato da cellule DH5α trasformate. Il DNA plasmidico è stato recuperato da cellule DH5α trasformate, digerite con HindIII, eseguite in un gel di acarosio all'1% e visualizzate con una fonte UV (passaggi da 2,19 a 2,22).
3. Analisi dei dati e risultati
Per calcolare l'efficienza di trasformazione, un indicatore di quanto bene le cellule hanno preso il DNA extracellulare, le colonie ottenute nella trasformazione devono essere contate:
| Diluizione | Cfu |
| 1/100 | 34 |
| 1/10 | 246 |
Tabella 1: Unità formanti colonie (cfu) conteggiate dall'esperimento di trasformazione.
L'efficienza di trasformazione (TE) è una misura del numero di cfu risultante dalla trasformazione di 1 μg di plasmide in un dato volume di cellule competenti. Molti parametri influenzano l'efficienza della trasformazione: dimensione del plasmide, genotipo cellulare, fase di crescita durante la preparazione della competenza, metodi di trasformazione, ecc.). Quando si calcola il TE è importante considerare quale diluizione (se presente) è stata eseguita prima della placcatura e incorporarla nel calcolo del numero totale di cfu. L'efficienza di trasformazione (TE) viene calcolata con la seguente equazione:

Per prima cosa dividi il cfu per il μg di DNA, in questo esempio 0,0001μg. Quindi dividere il risultato per il fattore di diluizione. In questo esempio, è stata utilizzata una diluizione di 1/10 e sono stati placcati 100μL di una soluzione da 1 ml (diluizione: 1/10 × 100 μL /1000 μL = 0,01).

I batteri sono notevolmente adattabili e un meccanismo che facilita questo adattamento è la loro capacità di assumere molecole di DNA esterne. Un tipo di DNA che i batteri possono assorbimento è chiamato plasmide, un pezzo circolare di DNA che contiene spesso informazioni utili, come i geni di resistenza agli antibiotici. Il processo di modifica dei batteri da parte di nuove informazioni genetiche incorporate da una fonte esterna è indicato come trasformazione. La trasformazione può essere facilmente eseguita in laboratorio utilizzando Escherichia coli, o E. coli.
Per essere trasformate, le cellule di E. coli devono prima essere rese competenti, il che significa in grado di assumere molecole di DNA dal loro ambiente. Il protocollo per realizzare questo è sorprendentemente semplice, una breve incubazione delle cellule in una soluzione di cloruro di calcio. Questa incubazione fa sì che le cellule diventino permeabili alle molecole di DNA. Dopo che le cellule sono state pellettizzate mediante centrifugazione, il surnatante viene rimosso. Il DNA plasmidico viene ora aggiunto alle cellule competenti. Dopo aver incubato le cellule con il DNA, la miscela viene brevemente riscaldata a 42 gradi Celsius, seguita da un rapido raffreddamento sul ghiaccio. Questo shock termico fa sì che il DNA venga trasferito attraverso la parete e le membrane della cellula. Le cellule vengono quindi incubate in mezzi freschi. Quindi, i batteri vengono posizionati a 37 gradi per consentire loro di risigillare le loro membrane ed esprimere proteine resistenti.
Quelle cellule che hanno assorbito i plasmidi copieranno fedelmente il DNA e lo passeranno alla loro progenie ed esprimeranno tutte le proteine che potrebbero essere codificate da esso, compresi i mediatori della resistenza agli antibiotici. Questi geni di resistenza possono essere utilizzati come marcatori selezionabili per identificare i batteri che sono stati trasformati con successo perché le cellule che non hanno assorbito il plasmide non esprimeranno il prodotto del gene di resistenza. Ciò significa che quando le cellule sono placcate su un terreno solido che contiene l'antibiotico appropriato, cresceranno solo le cellule che hanno assorbito il plasmide. La trasformazione delle cellule in una colonia in crescita può essere ulteriormente confermata coltivando quelle cellule in mezzi liquidi durante la notte per aumentare la resa prima di estrarre il DNA dal campione. Una volta isolato il DNA, è possibile eseguire un digest enzimatico di restrizione diagnostica. Poiché gli enzimi di restrizione tagliano il DNA in posizioni prevedibili, l'esecuzione di questi digest su un gel dovrebbe mostrare un modello prevedibile se il plasmide desiderato è stato trasformato con successo. Ad esempio, se pUC19 viene preparato e tagliato con l'enzima di restrizione HindIII, una singola banda di 2686 nucleotidi dovrebbe essere vista sul gel.
In questo laboratorio, trasformerai il ceppo di E. coli DH-5 Alpha con pUC19 e quindi confermerai la trasformazione riuscita mediante elettroforesi su gel di DNA.
Prima di iniziare la procedura, indossare i dispositivi di protezione individuale appropriati, tra cui un cappotto da laboratorio e guanti. Quindi, sterilizzare lo spazio di lavoro con il 70% di etanolo.
Ora, prepara cellule chimicamente competenti depositando un ciclo pieno di batteri su una piastra di agar LB sterile e strisciando i batteri con un nuovo anello. Quindi, incubare la piastra a 37 gradi Celsius durante la notte. Il giorno successivo, sterilizzare nuovamente il piano di lavoro con il 70% di etanolo e rimuovere la piastra dall'incubatrice.
Inoculare una singola colonia ben isolata in 3 millilitri di brodo LB in un tubo con un anello sterile. Quindi, coltiva la cultura a 37 gradi Celsius durante la notte, con agitazione a 210 RPM. Il giorno successivo, misurare la densità ottica della coltura notturna con uno spettrofotometro. Quindi, aggiungere 100 millilitri di brodo LB a un pallone da un litro e inocularlo con la coltura notturna a una densità ottica di 0. 01. Ora, incubare la coltura a 37 gradi Celsius con agitazione e controllare l'OD600 ogni 15-20 minuti fino a quando la coltura raggiunge la fase di crescita media-esponenziale.
Dopo circa tre ore, trasferire 50 millilitri della coltura in due bottiglie di polipropilene ghiacciato. Quindi, rimettere le bottiglie sul ghiaccio per 20 minuti per raffreddare. Quindi, recuperare le cellule tramite centrifugazione. Scartare i supernatanti e mettere le bottiglie a testa in giù su un tovagliolo di carta. Quindi, risuscisci il pellet batterico in cinque millilitri di soluzione di cloruro di magnesio cloruro di calcio ghiacciato e ruotare con attenzione fino a quando il pellet non si è sciolto completamente. Quindi, aggiungere altri 25 millilitri della soluzione al pellet batterico disciolto. Sospendare l'altro pellet batterico come precedentemente dimostrato. Dopo questo, ripetere la centrifugazione e rimuovere i supernatanti.
Se le cellule competenti stanno per essere trasformate direttamente, risusciere ogni pellet batterico in due millilitri di una soluzione ghiacciata di cloruro di calcio molare 0,1 facendo roteare attentamente i tubi. Per iniziare la procedura di trasformazione, trasferire 50 microlitri di celle competenti in due tubi in polipropilene da 1,5 millilitri etichettati. Quindi, aggiungere un microlitro di DNA plasmidico pUC19 a uno dei tubi. Mescolare delicatamente, evitando la formazione di bolle, e incubare entrambi i tubi per 30 minuti sul ghiaccio. Dopo l'incubazione, trasferire i tubi in un blocco di calore e incubare a 42 gradi Celsius per 45 secondi. Trasferire immediatamente i tubi sul ghiaccio e incubare per due minuti. Ora, aggiungi 950 microlitri di mezzi SOC a ciascun tubo e incubali per un'ora a 37 gradi Celsius per consentire ai batteri di recuperare ed esprimere il marcatore resistente agli antibiotici codificato nel plasmide.
Per effettuare una diluizione da 1 a 100, aggiungere 990 microlitri di mezzi SOC e 10 microlitri di sospensione cellulare in un tubo da 1,5 millilitri. Quindi, effettuare una diluizione da 1 a 10 aggiungendo 900 microlitri di mezzi SOC e 100 microlitri di sospensione cellulare a un tubo da 1,5 millilitri. Successivamente, piastra 100 microlitri delle sospensioni cellulari diluite e 100 microlitri del controllo negativo, su piastre selettive separate contenenti ampicillina usando uno spargitore e incubare le piastre a 37 gradi Celsius per 12-16 ore. Dopo l'incubazione, contare le unità formanti colonie, o CFU, per piastra, ottenute attraverso la trasformazione, e registrare questi dati. Per verificare che i trasformanti abbiano il plasmide pUC19, prelevare una singola colonia ben isolata da una piastra con un anello sterile e introdurla in un tubo contenente 3 millilitri di brodo LB. Quindi, incubare la cultura a 37 gradi Celsius con agitazione, durante la notte. Il giorno successivo, utilizzare un mini kit di preparazione del DNA per isolare il DNA da 3 millilitri della coltura, secondo le istruzioni del produttore. Dopo aver completato la mini preparazione del DNA, digerire 1 microgrammo di pUC19 purificato con un enzima di restrizione a 37 gradi Celsius per 1 ora. Ora, carica 20 microlitri di una scala a peso molecolare, 1 microgrammo di DNA plasmidico digerito e 1 microgrammo di DNA plasmidico non digerito in pozzetti consecutivi di un gel di agarosio all'1% contenente 1 microgrammo per millilitro di bromuro di etidio. Quindi, eseguire il gel per 1 ora a 95 volt. Infine, visualizza il gel con un illuminatore UV.
In questo esperimento, le cellule chimicamente competenti di E. coli DH5 Alpha sono state preparate utilizzando un adattamento della procedura del cloruro di calcio e quindi trasformate con il plasmide pUC19 per determinare l'efficienza di trasformazione. Per calcolare l'efficienza di trasformazione, utilizzare i conteggi CFU registrati per le diluizioni 1 su 100 e 1 su 10 e qualsiasi altra diluizione con conteggi di CFU compresi tra 30 e 300. In primo luogo, il conteggio dei CFU registrato, 246 in questo esempio, è diviso per la quantità di DNA, .0001 microgrammi qui, che è stata placcata. Quindi, questo numero viene diviso per il fattore di diluizione utilizzato per dare l'efficienza di trasformazione in CFU per microgrammo. In questo esempio, è stata utilizzata una diluizione da 1 a 10 e sono stati placcati 100 microlitri di una soluzione da 1 millilitro, dando un fattore di diluizione finale di 0,01. Nella corsia plasmidico non digerito, il DNA circolare può apparire come due o tre diverse bande di luminosità variabile. Questo perché il DNA circolare e non tagliato può esistere in diversi stati di conformazione, come supercoiled, cerchio aperto o più lineare, e ognuno di questi si muove attraverso il gel a velocità diverse. L'analisi della digestione del DNA plasmidico recuperato ha indicato che il plasmide utilizzato ha una dimensione prevista di DNA pUC19, 2.686 coppie di basi.
Subscription Required. Please recommend JoVE to your librarian.
Results
Sebbene il TE dipenda da molti fattori, i preparati cellulari competenti non commerciali, come questo, normalmente producono da10 6 a 107 trasformanti per microgrammo di plasmide. Pertanto, questa preparazione, con un TE = 2,46 x 108 cfu/μg, ha prodotto un TE ben oltre l'intervallo previsto. Sono disponibili protocolli aggiuntivi per la realizzazione di celle supercompetenti quando sono richieste maggiori efficienze di trasformazione per unadeterminata applicazione ( 13).
L'analisi della digestione del DNA plasmidico recuperato dalle cellule trasformate ha indicato che questo plasmide ha la dimensione prevista del DNA pUC19 (2686 bp).
Subscription Required. Please recommend JoVE to your librarian.
Applications and Summary
La trasformazione è un metodo potente per introdurre DNA esogeno nelle cellule batteriche che è la chiave per molte applicazioni di biologia molecolare in laboratorio. Inoltre, svolge un ruolo importante in natura consentendo alle cellule batteriche di scambiare materiale genetico che potrebbe comportare una maggiore variazione genetica e consentire l'acquisizione di diversi tratti benefici per la sopravvivenza in una vasta gamma di condizioni. Molti ceppi batterici codificano i geni necessari per la competenza naturale. Tuttavia, le condizioni in cui questi geni sono indotti sono ancora sconosciute. Sono necessarie ulteriori ricerche per determinare queste condizioni.
Subscription Required. Please recommend JoVE to your librarian.
References
- Croucher, N. J. et al. Rapid pneumococcal evolution in response to clinical interventions. Science. 331 (6016):430-434. (2011)
- Borgeaud, S. et al. The type VI secretion system of Vibrio cholerae fosters horizontal gene transfer. Science. 347(6217):63-67. (2015)
- Burmeister, A. R. Horizontal Gene Transfer. Evol Med Public Health. 2015 (1):193-194. (2015)
- Weston A, Brown MG, Perkins HR, Saunders JR, Humphreys GO. Transformation of Escherichia coli with plasmid deoxyribonucleic acid: calcium-induced binding of deoxyribonucleic acid to whole cells and to isolated membrane fractions. J Bacteriol. 145 (2):780-7. (1981)
- Dagert M, Ehrlich SD. Prolonged incubation in calcium chloride improves the competence of Escherichia coli cells. Gene. 6 (1):23-8. (1979)
- Asif A, Mohsin H, Tanvir R, and Rehman Y. Revisiting the Mechanisms Involved in Calcium Chloride Induced Bacterial Transformation. Front Microbiol. 8:2169. (2017)
- Panja S, Aich P, Jana B, Basu T. How does plasmid DNA penetrate cell membranes in artificial transformation process of Escherichia coli? Mol Membr Biol. 25 (5):411-22. (2008)
- Silhavy, TJ, Kahne D, Walker S. The Bacterial Cell Envelope. Cold Spring Harb Perspect Biol. 2 (5): a000414. (2010)
- Panja S, Aich P, Jana B, Basu T. (2008) Plasmid DNA binds to the core oligosaccharide domain of LPS molecules of E. coli cell surface in the CaCl2-mediated transformation process. Biomacromolecules. 9 (9):2501-9.
- JoVE Science Education Database. Basic Methods in Cellular and Molecular Biology. Plasmid Purification. JoVE, Cambridge, MA. (2018)
- Bergkessel M and Guthrie C. Colony PCR. Methods in Enzymology. 529: 299-309. (2013)
- Sambrook J and Russell DW. Molecular Cloning A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.Protocol 25 (1.116-118). (2001)
- Wirth R, Friesenegger A, Fiedler S. Transformation of various species of gram-negative bacteria belonging to 11 different genera by electroporation. Molecular & General Genetics. 216 (1): 175-7. (1989)