Note: To protect the fluorophore-conjugated antibodies from light, perform all steps in a bio-safety cabinet with the light off.
1. Antibody Staining Panel Preparation
- The antibody staining panel can be found in Table 1. Antibody concentration should be defined by titration with whole blood and using the same flow cytometry equipment and procedures that will be used to acquire the stained phenotyping samples.
- Once appropriate staining titers are determined, combine all antibodies into a single mixture in a lock-cap tube. Add flow wash buffer (Dulbecco’s PBS with 2% heat inactivated fetal bovine serum) to bring the total volume to 100 μl. Scale the mixture for the number of samples being stained. This mixture can be stored at 4 °C for up to eight weeks.
2. Staining
- If the blood collected is to be used for other purposes in addition to this assay, set an aliquot aside while more time-sensitive procedures are performed on the remaining blood. The aliquot can be stored at room temperature for up to 4 hr after venipuncture without significant cell loss.
- Verify that there is an intact bead pellet at the bottom of the Trucount tube and label the tube to identify the sample being stained. In HVTN clinical trials, the Laboratory Data Management System (Frontier Science and Technology Research Foundation; Amherst, NY) is used to label and track stained samples.
- Record the lot numbers and expiration dates of all reagents. Record the Trucount tube bead count number provided by the manufacturer on the bag of tubes; make sure that the lot number on the bag matches the lot number on the tube.
- Use reverse pipetting to accurately pipette 100 μl of whole blood into the Trucount tube, just above the metal retainer. Avoid smearing blood down the side of the tube.
- Using regular (forward) pipetting technique, pipette 100 μl of the mixed antibody staining panel (See Table 1) into the Trucount tube. Cap the tube and vortex at low speed for approximately 15 sec to mix. Visually inspect the tube to ensure that the bead pellet is fully dissolved.
- Incubate the Trucount tube for 15 minutes at room temperature (15-30 °C) in the dark.
- If necessary, dilute an aliquot of 10× FACS Lysing solution to 1X using diH2O. Add 900 μl 1× FACS Lysing solution to the tube.
- Cap the tube and vortex thoroughly at low speed for approximately 15 sec to mix. Push the cap down firmly into locking position on the tube and seal with laboratory film.
- Store the tube at -65 °C to -95 °C until the sample is ready for shipment to the central analysis lab or for analysis in house. Samples are stable at this stage for at least four weeks. If shipping or assaying immediately, this step can be omitted.
3. Shipping
Note: The following instructions utilize an insulated shipping system from Saf-T-Pak, Inc. specifically designed for shipping category B exempt biological substances according to International Air and Transport Association (IATA) regulations. If analyzing the samples in the same location as the staining occurred, go to section 4.
- Samples can be shipped immediately after staining or once they are frozen at -65 °C to -95 °C. Wrap each tube completely in foil and place in the stained specimen box. Place the stained specimen box inside a leakproof poly-bag with absorbent material.
- Place the leakproof poly-bag and contents into a Tyvek bag and seal with as little air as possible in the bag.
- Place the specimen package (specimen inside secondary packaging) inside the inner brown box.
- Place the inner brown box inside the Styrofoam chest, securing it into the indentation to prevent shifting.
- Fill the Styrofoam chest with dry ice (approximately 8 kg) and place the lid tightly on the chest.
- Securely tape the shipping box and ship as Biological Substance, Category B (UN3373) with the proper Dry Ice (UN1845) markings; follow IATA PI-650 instructions.
- Upon receipt, samples are stored at -65 °C to -95 °C until they are analyzed.
4. Thawing and Flow Cytometry Analysis
- Remove the stained sample from the freezer to thaw at room temperature in the dark before collecting on the flow cytometer. If collecting data on more than one sample, standardize the process for all tubes by staggering thawing so that the tubes are not sitting at room temperature for more than 1 hr each.
- Samples should be acquired using a four laser flow cytometer equipped with appropriate filters, such as the BD LSR II. Use standard cytometer calibration and fluorescence compensation methods for data collection 4.
Note: Do not set forward scatter or side scatter thresholds during collection 5. Trucount beads can fall below the lowest possible threshold setting for these parameters causing a subset of beads to not be accounted for during analysis. If required by the instrument to set a threshold, set the lowest possible Am Cyan channel threshold. Because CD45+ leukocytes stained with the panel will be Am Cyan positive, and Trucount beads also fluoresce in the Am Cyan channel, this should allow for all relevant data to be appropriately collected.
- Vortex the sample tube for 5 sec before loading on the flow cytometer. Trucount beads fluoresce highly in many channels. Gate the beads during collection by finding the population that is highly double-positive for PE Cy5 and APC (the two colors that most easily distinguish the beads from the cells may vary depending on instrumentation). Select the bead gate as your stopping gate, and record data until at least 20,000 beads are acquired.
- Analyze the data using appropriate software, such as FlowJo (Treestar; Ashland, OR). Figure 1 shows the gating scheme utilized for analysis of different leukocyte populations from a representative control blood draw.
5. Trucount Calculations
- Each Trucount tube contains a lyophilized pellet of fluorescent beads. After adding liquid to the tube and vortexing, the beads should become equally distributed throughout the sample. The number of beads in the pellet varies slightly by lot number and can be found on the storage bag for the tubes.
- Gate the Trucount beads and cell populations as shown in Figure 1 to determine event counts for each population. Comparing the number of events in the bead gate to the total number of beads originally in the tube will allow you to determine the ratio of sample collected, which can then be used to determine the absolute concentration (i.e., cells/μl) for each population. The following equation can be used for this purpose: Cell concentration (# cells/μl whole blood)= [# population events/(# bead events/# total beads in pellet)]/100 μl.
6. Representative Results
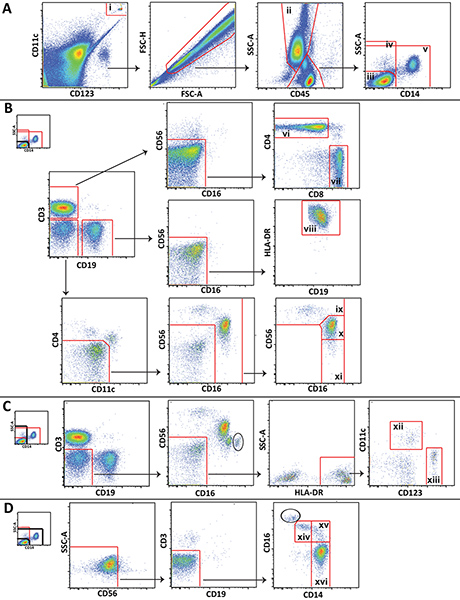
Figure 1. Gating scheme utilized for analysis of major leukocyte populations showing representative data from a healthy volunteer. A) Trucount beads (i) are gated and excluded from cells. Granulocytes (ii) are delineated and lympohcytes and monocytes are divided into 3 popluations: CD14 negative lymphocytes (iii), all CD14 negative cells (iv), and non-lymphocytes (v). B) CD14 negative lymphocytes are gated to distinguish CD4+ T cells (vi), CD8+ T cells (vii), B cells (viii), CD56 bright NK cells (ix), CD56 dim NK cells (x), and CD56 negative NK cells (xi). C) All CD14 negative cells are gated to distinguish myeloid (xii) and plasmacytoid (xiii) dendritic cells. D) Non-lympocytes are gated to distinguish non-classical (xiv), intermediate (xv), and classical (xvi) monocytes. Click here to view larger figure.
More specific cell subsets that are not shown in Figure 1 (e.g., NKT cells or neutrophils) can also be distinguished using the panel we present, and the gating scheme can be expanded or modified to meet specific study needs. Certain gating steps shown are unique to this method. Of particular note, an inclusion gate and exclusion gate are drawn around the Trucount beads and placed on top of one another, one to gate the beads for counting, and one to exclude the beads from the cellular analysis (Figure 1A). Also, because lymphocytes, monocytes, and granulocytes are not as easily distinguished in whole blood as they are in peripheral blood mononuclear cells by forward scatter and side scatter, gating these cells using CD45 expression and side scatter is often necessary (Figure 1A). Contaminating granulocytes (circled) that could not be separated from lymphocytes and monocytes using CD45 and side scatter are distinguishable in some plots by their high CD16 expression (Figure 1C and Figure 1D). The number of contaminating granulocytes is usually small, and they do not interfere with monocyte and NK cell gating.
