Three representative figures are shown here, including cytokine production in the lung of γHV68-infected Mavs+/+ and Mavs-/- mouse, cytokine secretion and gene expression level of γHV68-infected Mavs+/+ and Mavs-/- MEFs, and cytokine mRNA levels of γHV68-infected Mavs-/- MEFs "reconstituted" with MAVS. These representative experiments utilize gene knockout mice to investigate antiviral cytokine production in vivo and explore knockout MEFs to dissect the mechanism of regulated cytokine production ex vivo. Specifically, γHV68 infection of Mavs-/- mice led to significantly higher production of CCL5 in vivo. Mavs-/- MEFs produced more antiviral cytokine than Mavs+/+ MEFs during γHV68 infection, which recapitulates the in vivo phenotype. "Reconstituted" expression of MAVS in Mavs-/- MEFs decreased CCL5 production. Collectively, these results demonstrate that MAVS is necessary for γHV68 to suppress antiviral cytokine production both in vivo and ex vivo.
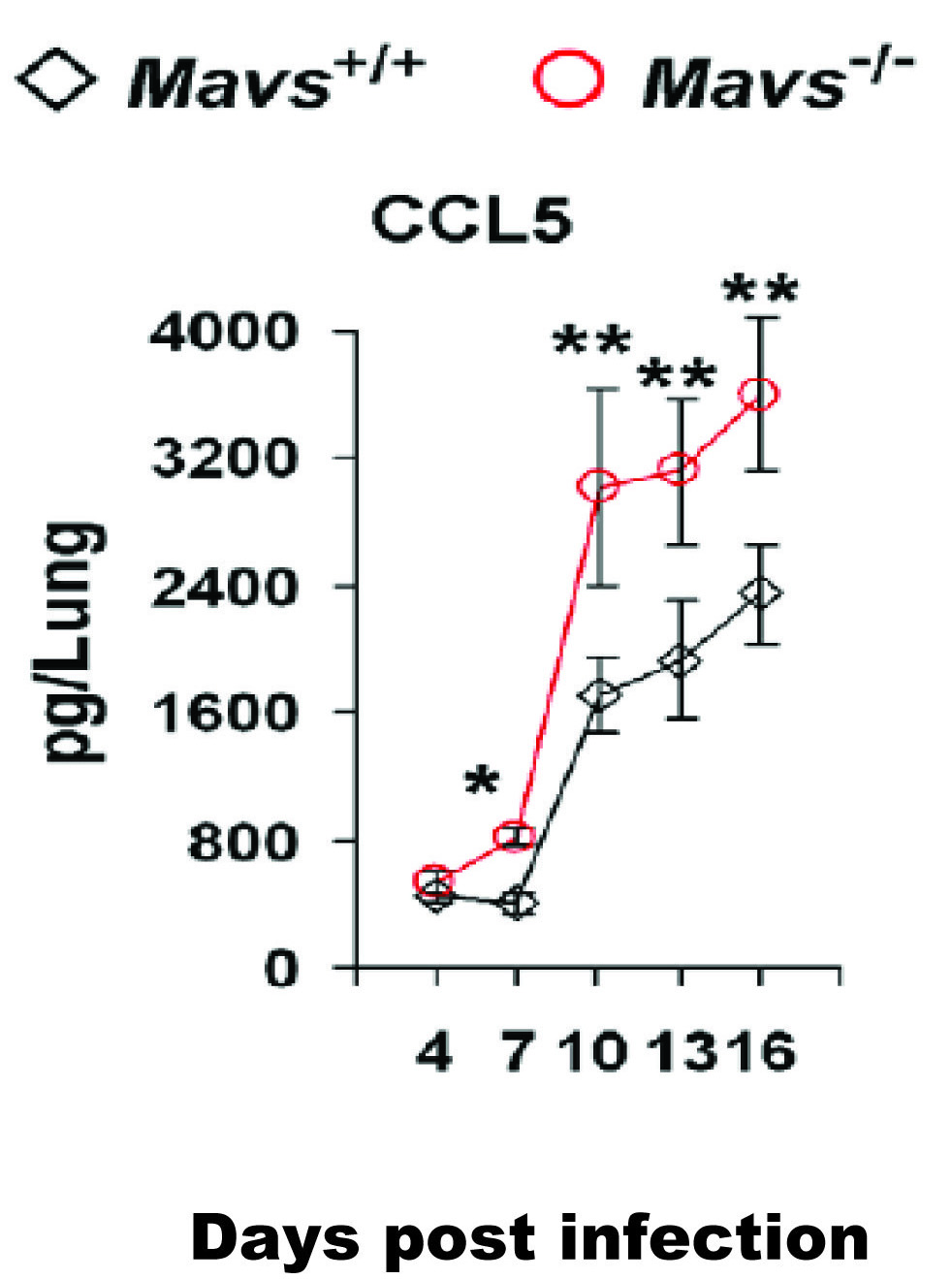
Figure 1. γHV68 acute infection induced higher cytokine production in the lungs of Mavs-/- mice than that in Mavs+/+ mice. Age- and gender-matched Mavs+/+ and Mavs–/- mice were intranasally infected with 40 PFU of γHV68 and CCL5 in the infected lungs at indicated days post-infection was measured by ELISA. γHV68 infection of Mavs-/- mice led to significantly higher production of antiviral cytokine CCL5, which suggested that MAVS is necessary for γHV68 to suppress antiviral cytokine production. Data are presented as the mean ± the standard error of the mean (SEM). The statistical significance: *,p<0.05; **,p<0.02. Click here to view larger image.

Figure 2. Upon γHV68 infection, Mavs-/- MEFs produced more antiviral cytokine than Mavs+/+ MEFs. Mavs+/+ and Mavs-/- MEFs were infected with γHV68 and harvested at indicated hours post-infection (h.p.i.), CCL5 in the supernatant was assessed by ELISA (A) and gene expression in MEF cells by quantitative real-time PCR (B). The results of Mavs-/- MEFs infected with γHV68 recapitulate those of γHV68-infected Mavs-/- mice. Data are presented as the mean ± SEM of three independent experiments. The statistical significance: **,p<0.02, ***,p<0.005. Click here to view larger image.

Figure 3. "Reconstituted" expression of MAVS in Mavs-/- MEFs decreased CCL5 production. (A) Mavs-/- MEFs were infected with control or MAVS-expressing lentivirus and the expression of MAVS was analyzed by immunoblotting. (B) Mavs-/- MEFs "reconstituted" with MAVS were infected with γHV68. MEFs were harvested at the indicated hours post infection (h.p.i.) and RNA was analyzed by quantitative real-time PCR with primers specific for CCL5. Data are presented as the mean ± SEM of three independent experiments. The statistical significance: **,p<0.02. Click here to view larger image.
