Enhanced Northern Blot Detection of Small RNA Species in Drosophila Melanogaster
Summary
The aim of this publication is to visualize and discuss the operative steps of an Enhanced Northern Blot protocol on RNA extracted from Drosophila melanogaster embryos, cells, and tissues. This protocol is particularly useful for the efficient detection of small RNA species.
Abstract
The last decades have witnessed the explosion of scientific interest around gene expression control mechanisms at the RNA level. This branch of molecular biology has been greatly fueled by the discovery of noncoding RNAs as major players in post-transcriptional regulation. Such a revolutionary perspective has been accompanied and triggered by the development of powerful technologies for profiling short RNAs expression, both at the high-throughput level (genome-wide identification) or as single-candidate analysis (steady state accumulation of specific species). Although several state-of-art strategies are currently available for dosing or visualizing such fleeing molecules, Northern Blot assay remains the eligible approach in molecular biology for immediate and accurate evaluation of RNA expression. It represents a first step toward the application of more sophisticated, costly technologies and, in many cases, remains a preferential method to easily gain insights into RNA biology. Here we overview an efficient protocol (Enhanced Northern Blot) for detecting weakly expressed microRNAs (or other small regulatory RNA species) from Drosophila melanogaster whole embryos, manually dissected larval/adult tissues or in vitro cultured cells. A very limited amount of RNA is required and the use of material from flow cytometry-isolated cells can be also envisaged.
Introduction
RNA biochemistry has experienced spectacular progresses in the past years1. Our comprehension of RNA potential in the control of gene expression has been burst by the identification of powerful noncoding ribo-regulators2, by the discovery of novel RNA-based regulatory mechanisms3,4 and by a deeper characterization of already known post-transcriptional events5. All together these studies have allowed RNA biology to dramatically make the scene, becoming a major research subject in the current scientific landscape. In particular, over recent years we are getting a sense of the pervasive impact of the “RNA world” on molecular neurobiology6, one of the most dynamic research domains in modern life science. In the last decade of the past century, the overall scientific scenario has been revolutionized by the discovery of the RNA interference7,8 and of the small regulatory RNAs9,10 with particular regard to microRNAs11, endogenously expressed small noncoding RNAs implicated in the control of nearly all cellular functions as pleiotropic and combinatorial regulators of gene expression.
Almost 10 years after miRNA initial discovery in Caenorhabditis elegans by Ambros and Ruvkun’s labs, renewed attention was turned to the field when high numbers of miRNAs were identified in Drosophila and in human cells as well12-15. Since then, thanks to versatile transgenic approaches, Drosophila melanogaster has stood out as a valuable biological context for delving into miRNA biosynthesis and activity. Drosophila miRNAs have revealed distinct functions in insect-specific or evolutionarily conserved processes, spanning from aging to metabolism, signaling pathways, behavior and, of course, neurogenesis. Along this direction, we recently unveiled a novel link16 in the intriguing correlation occurring between the master gene gcm/glide and the RNA pathway. The fly transcription factor Gcm/Glide17-19 constitutes a unique example of cell fate determinant, which dictates the glial vs. neuronal fate choice in multipotent fly neural precursors20. Twenty-year long research on this topic has clearly underlined the occurrence of multiple and overlapping inputs of gene expression regulation converging over gcm/glide21-28 to establish the threshold levels required for balancing the delicate ratio between neuronal and glial counterparts during neural development.
We discovered that regulation via Dmel-miR279 targeting represents a further control level contributing to post-transcriptional fine-tuning of gcm/glide expression16. Globally, these research lines have required specific methodological improvements: along years, several technologies originally developed to analyze traditional RNAs have been converted for quantifying small non coding RNAs, like as RNAse protection assays, cDNA arrays29-31, real-time PCR methods32-35 and sequencing36,37. On the other side, recalibration of technical approaches has fostered continuous progresses in the field.
Northern Blot assay (NB, or RNA gel blot assay) constitutes a representative instance: it is largely employed to profile RNA accumulation, since it ensures both expression level quantitation as well as size determination. However, the intrinsic poor sensitivity of the method is limiting when it is to be applied to low-abundant gene expression fine tuners, like short RNAs. A detrimental consequence is the requirement of large amounts of total RNA, which makes difficult its application to specific biological samples. For such reasons, specific NB variants for small RNAs detection have been developed38-40: we took advantage of an improved NB procedure41 (ENB, Enhanced Northern Blot), while elucidating the abovementioned interplay between Dmel-miR279 and gcm/glide.
This method relies on a chemical crosslinking step based on the activity of a Carbodiimide derivative [1-ethyl-3-(3-dimethylaminopropyl)carbodiimide, EDC] to fix nucleic acid onto solid supports. Carbodiimide is a versatile cross linker known to catalyze the formation of amide bonds between activated carboxyl or phosphate groups and amine groups42. This property can be exploited to covalently couple small RNAs via their mono-phosphorylated 5′-hydroxyl group to amino groups at the surface of nylon membranes. The resulting attachment configuration increases the accessibility of the immobilized nucleic acid and, in turn, the efficiency of probe-target hybridization, which results in remarkable detection enhancement43.
The technique assumes particular relevance in Drosophila molecular studies, by which the occurrence of novel and distinctive classes of small noncoding RNAs is emerging44. Among these, rasiRNAs45,46 constitute a specific subtype of piwi-interacting RNAs (piRNAs47), involved in sequence-specific gene silencing. Operative details of this method are fully described and visualized hereinafter, relative to the analysis of the microRNAs Dmel-miR279 and Dmel-miR286 and, for the first time, of one rasiRNA, rasi4. We pushed to extremes this method, which allowed us revealing poorly expressed targets from minimal amounts of RNA (less than 1 μg).
Protocol
1. Sample Collection
- Detach semi-adherent Schneider’s 2 cells, (1-5 x 106 cells on average), by pipetting and harvest them by centrifugation (100 x g for 1 min). In case of in vitro cultured adherent cells, trypsinize them in standard conditions or directly collect them from plates into a convenient amount of RNA extraction reagent, by help of a cell scraper.
- For embryo collection, grow Drosophila strains in fly cages and let embryos accumulate on egg laying plates. Wipe embryos off by a paintbrush in distilled water (dH2O), discard debris by sieve filtering, rinse and recover embryos in a vial48.
- As an alternate technique, manually dissect tissues/organs. Isolate testes (a preferential system for piRNA analysis) from Drosophila abdomen in phosphate buffered saline (PBS) under a stereomicroscope (20X magnification), by using dissection needles or forceps as visualized in Sullivan et al49.
- Pestle homogenize samples upon addiction of 0.5 volumes of RNA extraction reagent.
2. RNA Extraction/Analysis
- Following manufacturers’ instructions perform RNA isolation through commercial reagents. CAUTION! RNA extraction agents are usually toxic and caustic. Alternatively, apply a Proteinase K-based protocol for RNA isolation50, as follows:
- Dilute samples into 400 μl of Stop Mix and incubate at RT for 15 min.
- Recover hydrosoluble components by standard phenol:chloroform:isoamyl alcohol and chloroform extractions and by ethanol (EtOH) precipitation/washing. Air dry the samples and resuspend into diethylpyrocarbonate (DEPC)-treated double distilled (dd)H2O.
- Assess RNA concentration by UV spectrophotometric measurement. Also ensure the RNA purity is high, close to 2.0, based on A260/A280 reading.
- Check RNA preparation on denaturing agarose gel as follows:
- In a clean beaker, dissolve 1.2 g of Agarose in 82 ml of DEPC-ddH2O, under continuous stirring. Boil until completely dissolved.
- Allow the gel solution to cool down; when temperature reaches 60 °C add 10 ml of 10X 4-morpholinepropanesulfonic acid (MOPS) buffer to 1X final concentration (FC), and 8 ml of 37% formaldehyde to 3% FC. CAUTION! Formaldehyde is a carcinogen.
- Pour the gel into a horizontal electrophoresis cell. Allow the gel to solidify under a hood for at least 1 hr. Following solidification, submerge the gel into MOPS 1X.
- Aliquot 2 μg of RNA sample and 1 μg of Ladder (use an RNA marker when accurate evaluation is desired). Add 3 volumes of agarose loading dye (ALD) to RNA and to Ladder. Heat at 70 °C for 5 min and immediately cool on ice. CAUTION! Contents of ALD (ethidium bromide and formamide, see Materials) are mutagen and corrosive, respectively.
- Load the samples and run the gel at 50 V for about 1 hr with occasional buffer recycling. Check the RNA fractionation pattern by UV visualization or gel imaging. Recognize discrete bands corresponding to 28S and 18S rRNAs and to tRNAs (see Figure 1).
- Proceed to Northern Assay or store RNA at -80 °C.
3. Denaturing Acrylamide Gel Preparation
- Use a small vertical electrophoretic apparatus for this fractionation step (plate size about 8 cm x 9 cm, comb thickness 0.75 mm).
- Assemble clean glasses together in a casting frame.
- Prepare 10 ml of 10% acrylamide/bis-acrylamide (19:1) solution in MOPS 1X, 7 M urea. Immediately before pouring, add 100 μl of 10% ammonium persulfate and 10 μl of TEMED and rapidly mix the solution. CAUTION! Acrylamide is neurotoxic and carcinogen.
- Pour the gel between glass plates and insert the well comb carefully to avoid bubbles. Let the gel polymerize at RT for at least 45 min before use. Alternatively, store the gel O/N, wrapped in water-soaked filter papers and sealed in saran wrap.
4. Sample Preparation
- Aliquot 0.5-20 μg of total RNA for each sample. If volume exceeds 3-4 μl, vacuum dry the sample.
- For size determination, run an aliquot (20-30 cps) of radiolabeled low-range ladder in parallel to samples, as a marker (see section 4.3 and 4.4 for preparation). Treat it in parallel to samples, as described in step 4.5.
- Ladder: set up a phosphate forward reaction using 0.1 μg of a 10 bp-step ladder. Incubate at 37 °C for 30 min and stop the reaction by adding ethylenediaminetetraacetic acid (EDTA) to 20 mM FC.
- Purify by EtOH precipitation/washing, air dry and resuspend in ddH2O (about 100 μl). CAUTION! 32P is a radioactive source.
- To each sample add an equal volume of acrylamide blue dye (ABD). Denature by heating at 80 °C for 5 min and cool on ice until loading. CAUTION! Formamide (content in ABD) is corrosive.
5. Electrophoretic Fractionation
- Remove the glass plates from the casting support and properly assemble them in the electrophoresis cell. Fill both inner and outer chamber with sufficient Running Buffer (RB).
- Gently remove the comb and pre-run the gel at 200 V for 10 min. Before loading samples, wash away urea deposits from the wells by squirting some RB through a syringe.
- Use flat tips to carefully stratify samples from step 4.5 at the bottom of each well. Check anode and cathode connections to avoid inverted running. After the samples enter the gel at 100 V for 5 min, increase the voltage to 200 V.
- For a fractionation length of about 7 cm, a 45 min-long run is sufficient. Avoid Bromophenol blue (a tracking pigment in ABD) to overflow the gel.
- Visualize on-gel the fractionated long RNAs by ethidium bromide (EtBr) staining, as an optional step:
- Cut away 2 cm from the top of the gel and briefly rinse the slice in ddH2O.
- Incubate the gel slice at RT for 15 min with gentle shacking in RB, EtBr 0.5 μg/ml FC.
- Destain the gel for 15 min in RB, visualize long rRNAs by UV irradiation or gel imaging. Check for RNA loading and quality (see Figure 1, Panel 7). CAUTION! Ethidium bromide is a mutagen.
- Meanwhile proceed blotting the lower part of the gel containing small RNA species.
6. Gel Blotting
- Cut 6 pieces of blotting paper to fit the blotting area and an equivalent piece of uncharged nylon membrane. Dampen paper sheets, membrane and 2 blotting pads in RB. Make sure to completely soak the pads by repeatedly squeezing them in RB.
- Remove the gel from the apparatus and open the glasses by mean of a spatula. Use a clean cutter to remove the wells. Place a sheet of wet Whatman paper on the gel and gently lift it up.
- Place the nylon membrane on the free face of the gel. Mark a corner to orient the filter according to sample loading. Complete the “sandwich” (3 paper pieces each side). Roll a plastic rod on the blot surface, to remove any possible air bubbles.
- Place the blot between two blotting pads and then in the blotting cassette. Assemble the cassette in the blotting module, fill the chamber with RB and check for the correct orientation (nylon membrane must stay between the gel and the positive terminus).
- Transfer at 20 V for 20 min. Note: to ensure homogeneous conditions, after 10 min open the blotting module and rotate the sandwich by 180° before completing the transfer.
7. Membrane Crosslinking
- Prepare 6 ml of fresh Crosslinking Solution (XLS). Saturate a 10 cm x 10 cm (larger than nylon membrane) piece of blotting paper with XLS. CAUTION! HCl (a component of XLS) is highly corrosive.
- Dismantle the blot and place the membrane on the wet 3MM filter paper. Note: RNA must not be in direct contact with the saturated paper. Place the filter and paper between two glass plates and wrap in saran-wrap.
- Heat the membrane at 60 °C for 2 hr, followed by a ddH2O washing. Store at -20 °C or proceed to hybridization.
8. Membrane Prehybridization
- If RNA loading has been checked (optional step 5.5), directly proceed to hybridization. Or else, cut away 1 cm from to the top of the nylon filter, to avoid probe cross-hybridizations to fractionated long RNAs.
- Preheat 10 ml of Hybridization Solution (HS) at 37 °C. Denature 1 mg of Salmon Sperm at 95 °C for 5 min, cool on ice and add to HS.
- Incubate the filter in HS at 37˚C for at least 1 hr, under rotation in a hybridization oven. Check that the RNA-side of the filter faces HS.
9. Probe Synthesis
- Set up a phosphate forward reaction on 10 pmol of specific antisense oligonucleotide, as described in 4.3.
- Add 5 volumes of Tris-EDTA-NaCl (TEN) buffer to the reaction and purify the labeled primer from unincorporated nucleotides by gel exclusion chromatography on G-25 Sephadex columns (or also by EtOH precipitation). Probe activity can be assessed by Liquid Scintillation Counter.
10. Membrane Hybridization
- Replace the exhausted HS with a fresh 10 ml aliquot (prepared and complemented as described above). Heat the probe at 95 °C for 10 min, cool on ice and add to novel HS. Incubate the filter at 37 °C ON.
11. Membrane Washing
- Recover the hybridization solution and store it at -20 °C in a radioactivity-shielding box.
- Rinse the filter in Washing Buffer (WB) and wash it thoroughly for 10 min at RT.
- Use a Geiger Mueller Detector for checking residual radioactivity at the filter corners. When background emission is around 5 cps, proceed to membrane exposition.
12. Signal Detection
- Expose the filter on autoradiography films or on a molecular imager screen ON. Reveal signals by photographic processing or digital conversion.
- Analyze the band intensity by dedicated quantification software (ImageJ or Optiquant).
13. Membrane Stripping
- To remove primary signals, submerge the membrane in 500 ml of boiling Stripping solution, then incubate at RT for 1 min. Proceed to novel (pre)hybridization or store the filter at -20 °C.
Representative Results
By following the overall procedure described in the text and schematically represented in Figure 1, we assessed small RNAs expression from complex RNA samples of different origins. In the experiment presented in Figure 1, RNA was isolated from Drosophila Schneider’s 2 cells by Proteinase K extraction, checked for integrity (optional step: denaturing agarose gel fractionation) and loaded in double. One half of gel was blotted on a neutral membrane and chemically cross-linked by EDC; the second half was transferred on a positively charged nylon filter, then UV cross-linked (1.2 x 105 μJ/cm2). The endogenous expression of two microRNAs belonging to the Dmel-miR279 family (Dmel-miR279 itself and Dmel-miR286 15) was verified in both conditions. The improved procedure (ENB) ensures higher sensitivity respect to the standard one (SNB).
The different efficiency between the two methods is even better highlighted in Figures 2 and 3. The experimental rationale is the same as above: here we report the representative result of Northern Blot autoradiographic detection of the piRNA rasi4 along a titration of total RNA extracted from adult fly testes. ENB already allows detecting a specific band when 0.5 μg of RNA is loaded. The quantification aside shows how the signal proportionally increases with the amount of fractionated RNA, indicating that the dose/response ratio lies in linear range of detection. The panel below ratifies the improved sensitivity of this method compared to SNB. In this case, a detectable signal is obtained upon electrophoretic fractionation of 10 μg of RNA at least for the SNB.
Similar results are obtained with a different noncoding RNA species, e.g., 5s rRNA, as reported in Figure 3.
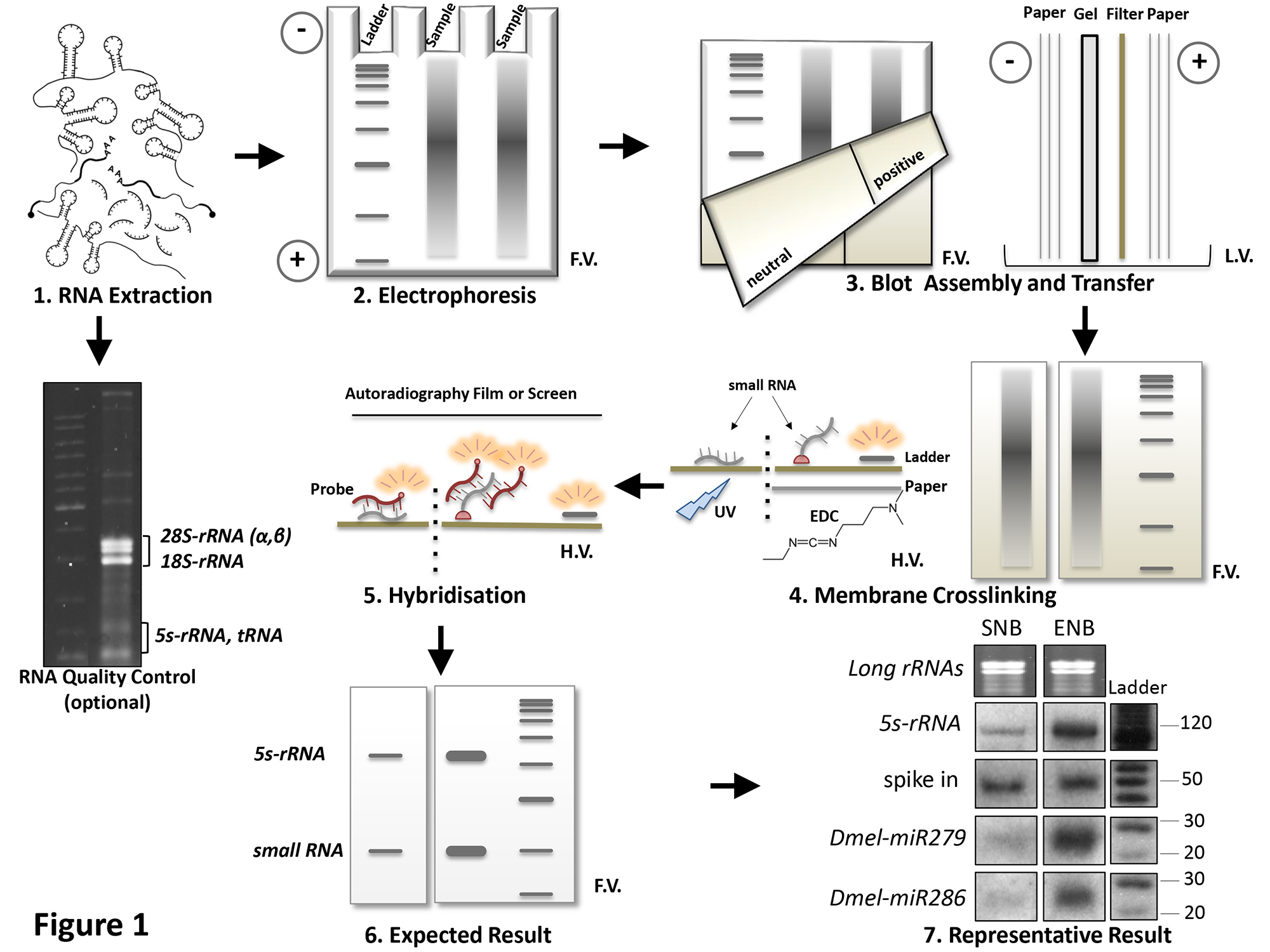
Figure 1. Schematic representation of the overall Northern Blot procedure. Main procedural steps are listed and progressively numbered. Results are compared between the Enhanced Northern Blot (ENB) and Standard Northern Blot (SNB), performed in same conditions: RNA isolation from Drosophila Schneider's 2 cells (Panel 1), electrophoresis (Panel 2), transfer (Panel 3) and hybridization (Panel 5). ENB refers to the improved procedure described in this report, which requires a neutral filter (Panel 3, left half of the membrane) and EDC-based crosslinking (Panel 4) which establishes a covalent binding between target RNA 5’-end and membrane (pink dots in Panel 4). In SNB, RNA crosslinking is obtained by UV treatment (Panel 4) on a positively charged nylon membrane (Panel 3, right half of the membrane). 5s RNA specific signal is not a calibrator in comparative conditions, because it also responds to differences between SNB and ENB procedure. Thus, an amount (corresponding to 10 cps) of a 5’-labeled (see points 4.3 and 4.4 of the protocol), 50 nt long scrambled oligonucleotide was added to each sample before electrophoresis, to function as a “spike in” reference (Panel 7). Loading can be also assessed by on-gel EtBr staining of long rRNAs (see point 5.5 of the protocol). L=Ladder. F.V.=Frontal View, L.V.= Lateral View, H.V.= Horizontal View. Please click here to view a larger version of this figure.

Figure 2. Comparative Northern Blot analysis of rasi4 expression. Endogenous levels of the testis-specific piRNA rasi4 are revealed by enhanced (ENB) or standard (SNB) Northern Blot, performed on increasing amounts of total RNA (indicated above each lane) extracted from adult Drosophila testes, or from whole abdomens (Abd). For each methodology, histograms aside report the relative amounts of rasi4 respect to the same amount of a “spike in” loading control. Quantifications from the two methods are combined in the conclusive graph placed below the gels. Please click here to view a larger version of this figure.

Figure 3. Comparative Northern Blot analysis of 5s rRNA expression. As a positive control and experimental calibration, previous analysis was extended to 5s rRNA. Details as in Figure 2. Please click here to view a larger version of this figure.
Discussion
Although our appreciation of the sophisticate intertwining through which RNA regulates neurogenesis is continuously increasing, the mechanistic implications of RNA-based circuitries affecting neural stem cell biology, neuronal differentiation/functional integration, development of neural pathologies and cancers remain unexplored. The maintenance of such networks through kingdoms makes the potential of genetic systems as Drosophila melanogaster instrumental to unravel unexplored cellular pathways, in which short noncoding RNAs and transcription factors are regarded as major molecular players. In this view, the profiling of candidate small RNA expression levels is propaedeutic to accurate functional analyses: an example comes from our recent demonstration that Dmel-miR279 modulates the gcm/glide fate determinant in vivo, accomplished through an improved version of Northern Blotting assay as a tool for expression analysis.
Since its initial development in 197751, NB has been regarded as a reference method for RNA analysis. In spite of some intrinsic limitations of this assay, as the difficult conversion to multiplex format or possible errors with signal quantification, recent breakthroughs in RNA molecular biology have rediscovered this conventional technique as a “quick and clean” system for assaying endogenous expression of small ribonucleic acids or for validating the effectiveness of gain- or loss-of-function approaches. As paradigmatic instance, Northern analysis lays at the basis of the original discovery of microRNAs, whereas the approach remains at the forefront for its relevance in the characterization of ever-new properties of regulatory RNAs52.
The variant described here is based on increased sensitivity due to chemical crosslinking of RNA. We believethe method is still poorly employed in Drosophila, but when exploited led to significant discoveries52, since it makes possible profiling RNA expression from small samples (less than 1 μg), with an average 10-30 fold improvement of RNA target detection41,43, as we obtained in our conditions (Figures 2 and 3).
RNA integrity constitutes a critical issue in order to obtain consistent and reproducible results. RNA manipulations require accuracy and rapidity to limit possible sample degradation occurring between extraction and loading. On this purpose, all solutions need to be diethylpyrocarbonate- (DEPC) treated before use (2 hr incubation in the presence of 0.1% DEPC at 37 °C and then autoclaved) to circumvent RNase activity. Solution treatment guarantees better results respect to using DEPC water as a solvent. All equipment will be equally rinsed in DEPC ddH20, after cleaning by an RNase-specific surface decontaminant.
RNA Isolation: Different protocols can be used to isolate intact RNA. Any commercial Guanidinium thiocyanate-phenol-chloroform reagent can be successfully employed: they are highly recommended when small RNA expression is particularly low (e.g., rasi4, Figure2) or when RNA purification is not performed on fresh samples (for instance, preserved into tissue storage reagents). From experience, RNA isolation from stabilized samples resulted significantly harsher and required more drastic extraction conditions. In standard conditions, the described Proteinase K-based method guarantees good-quality preparations as well, (a significant example is represented in Figure1) but it is not the optimal choice for samples preserved in storage reagents.
Finally, since this procedure aims to small RNA specific detection, dedicated column purification-based kits can be employed for direct small RNA isolation, even though this makes harder to verify RNA integrity. In alternative to the classical gel-based procedure for RNA integrity control described above, RNA quality can be evaluated by instruments for on-chip miniaturized analysis of nucleic acid patterns.
Buffer Choice: MOPS-NaOH buffer (pH 7) was proposed as a preferential buffer for both Enhanced Northern gel electrophoresis and blotting. We also satisfactorily used Tris-borate-EDTA (TBE) buffer in our experiments. In this case, since the presence of nucleophile amine groups of Tris(hydroxymethyl)aminomethane would hydrolyze and inactivate DEPC during treatment itself, it is recommended dissolving Tris into DEPC-treated water instead of treating TBE buffer already prepared.
Electrophoretic Run and Blotting: Pre-running the gel is essential for removing excess Persulfate and eliminating hyperfocusing. Pre-run parameters should be maintained also during fractionation, avoiding too fast electrophoretic run. Before loading, make sure to remove possible Urea sediments from wells, by squirting buffer inside. This will limit sample spill-over and guarantees accurate RNA stratification, producing resolved autoradiographic bands during signals detection. Any neutral nylon membrane can be used as a support for nucleic acid transfer. Do not blot the RNA for more than 20-30 min at the specified conditions.
Crosslinking: EDC-based chemical crosslinking represents the critical methodological advance of this enhanced Northern blot version. The possibility to crosslink short RNA species to neutral surfaces by covalent bounds between the RNA 5’mono-phosphate group and nylon primary amines leaves the majority of bases available for hybridization. This enhances the sensitivity by 10-20xrespect to traditional crosslinking. It is crucial to prepare fresh amount of crosslinking solution for each blotting, in sufficient quantity for proper membrane saturation.
Hybridization and Washing: Probes should preferably be freshly labeled. Probe specific activity (dependent on the specific activity and amount of incorporated radiolabeled nucleotide and also on the amount of probe available for hybridization) determines the sensitivity of target detection. When [γ-32P] ATP with a specific activity of 3,000 Ci/mmol is used, 106 cpm/pmol for 100% labeling of the 5’ ends is expected.
The hybridization solution can be recovered and re-utilized, considering that, due to 32P decay, probe activity halves in about 14 days.
Due to membrane neutrality, low background noise is expected upon hybridization. If more stringent washing conditions are required, progressively reduce washing buffer ionic strength (up to 0.2x SSPE) or add increasing amounts of ionic detergents (up to 0.5% SDS) or raise washing temperature to 37°C.
To avoid overlapping signals with next hybridization, it is recommended to verify by re-exposing that the primary probe has been completely removed from the filter upon stripping. However, due to progressive loss of RNA from the filter more than 3-4 cycles of stripping and re-probing should be avoided.
Northern blot analysis is essential when RNA size measurement is required. Even though this assay may lack the accuracy of fluorescence-based qPCR approaches for estimation of tiny RNAs, an advantage derives from using denaturing fractionation/blotting and probe-dependent hybridization as unique steps before detection of target RNAs. Consequently, revelation of specific signals from the sample is direct and does not require any additional step of enzymatic treatment/conversion/amplification, which would imply the use of dedicated commercial kits. RNA levels can be quantified and compared between multiple samples on single membranes. Thus Northern blot is commonly employed as a stringent validation for qPCR data. Spurred by specific experimental requirements, the method has been even improved during the last years, both at level of sensitivity and resolution so that limited amount of RNAs (see Figures 2 and 3) are required for successful analysis.
The protocol we presented can be adapted to a wide range of applications especially where material is available in limited amounts. The biological source of RNA samples can be heterogeneous: as far as Drosophila is concerned, embryos or other developmental stages are suitable, but also manually dissected tissues or organs, individual cells sorting-isolated cytotypes or cell cultures.
Furthermore, even though microRNAs are the most abundant class of short regulatory RNAs in the cell and major subject of investigation in the field of RNA-mediated gene expression control, they do not constitute the only family of cellular noncoding RNAs. Many clusters of tiny RNAs function across kingdoms (e.g., endogenous siRNAs, piRNAs, snoRNAs, plant tasiRNAs) and most of them are characterized from a free 5’ terminus (unmodified), generally deriving from the endo-ribonucleolytic processing of precursors molecules. This feature constitutes the unique structural prerequisite strictly required for applying the specific improved methodology based on chemical crosslinking, even though RNA size have to be considered as well, when longer RNAs are analyzed. In Figure 2 we report a Northern Blot analysis comparatively detecting the levels of rasi454 a member of a class of germ-line-specific small RNAs (Piwi-interacting RNA or piRNAs.) In conditions of poor and specific expression levels, the enhanced sensitivity of this method can sanction successful results, especially considering that the reported protocol can be further improved by combining it with other practical implementations already described in literature55,56.
Disclosures
The authors have nothing to disclose.
Acknowledgements
This work was supported by the Institut National de la Santé et de la Recherche Médicale, the Centre National de la Recherche Scientifique, the Université de Strasbourg, the Hôpital de Strasbourg, the Association pour la Recherche sur le Cancer, the Institut National du Cancer, the Agence Nationale de la Recherche and the Région Alsace.
Pietro Laneve has been supported by the Fondation pour la Recherche Médicale. Currently, he is a recipient of a Istituto Italiano Tecnologia (IIT) fellowship. Publication costs are supported by the Neurex network (TriNeuron – Program Interreg IV Upper Rhine)
Materials
| FINAL COMPOSITION/NAME | COMPANY | CATALOGUE (Stock) | COMMENTS |
| GENERAL REQUIREMENT | |||
| Centrifuge, 5418 | Eppendorf | 5418 000.017 | |
| Decontaminant, RNase Away | Sigma-Aldrich | 83931 | Apply on glass/plasticware, wipe and rinse |
| RNase inhibitor, DEPC | Sigma-Aldrich | D5758 | Hazardous //// 1609-47-8 |
| 0.1 % DEPC suspension | Incubate 2 hr at 37 ˚C, then sterilize by autoclave | ||
| SAMPLE PREPARATION | |||
| Stereo Microscope, M60 | Leica | ||
| 1X PBS Buffer | |||
| 137 mM NaCl | Sigma-Aldrich | S3014 | 7647-14-5 |
| 2.7 mM KCl | Sigma-Aldrich | P9541 | 7447-40-7 |
| 10 mM Na2HPO4 | Sigma-Aldrich | S3264 | 7558-79-4 |
| 1.8 mM KH2PO4 | Sigma-Aldrich | P9791 | 7778-77-0 |
| RNA EXTRACTION/ANALYSIS | |||
| UV-Vis Spectrophotometer | Thermo Scientific | Nanodrop 2000 | |
| Gel Imager, ChemiDoc XRS+ | Biorad | 170-8265 | |
| Horizontal Electrophoresys, Mini-Sub Cell GT | BioRad | 170-4487EDU | |
| RNA extraction reagent, TRIzol Reagent | Life Technologies | 15596026 | Hazardous |
| PCA (25:24:1), 1:1 for extraction | Sigma-Aldrich | 77617 | 136112-00-0 |
| Chlorophorm, 1:1 for extraction | Sigma-Aldrich | C2432 | 67-66-3 |
| EtOH, 2.5 vol. for precipitation, 70% for washing | Sigma-Aldrich | 59844 | 64-17-5 |
| Gene Ruler 1Kb DNA Ladder | Thermo Scientific | SM0311 | |
| Stop Mix | |||
| 300 mM NaOAc, pH 5.5 | Sigma-Aldrich | S2889 | 127-09-3 |
| 1x RSB Solution | |||
| 2% sodium dodecyl sulfate (SDS) | Sigma-Aldrich | 71736 | Never cool down SDS to avoid precipitation //// 151-21-3 |
| 2 mg/ml Proteinase K | Roche Applied Science | 0-3115828001 | EC 3.4.21.64 9 |
| 10X Reticulocyte Standard Buffer (RSB) | |||
| 100 mM Tris-Cl, pH 7.4 | Sigma-Aldrich | T5941 | 1185-53-1 |
| 100 mM NaCl | Sigma-Aldrich | S3014 | 7647-14-5 |
| 30 mM MgCl2 | Sigma-Aldrich | M8266 | 7786-30-3 |
| Denaturing Agarose Gel | |||
| 1.2% Agarose | Sigma-Aldrich | A9539 | Let the gel solidify for at least 1 hr //// 9012-36-2 |
| 1x MOPS Buffer | Add when T < 60 ˚C | ||
| 3% formaldehayde | Sigma-Aldrich | F8775 | Hazardous. Add when T < 60 ˚C //// 50-00-0 |
| 10X MOPS Buffer | |||
| 0.2 M MOPS sodium salt, pH 7 | Sigma-Aldrich | M9381 | Do not confuse MOPS sodium salt with MOPS //// 71119-22-7 |
| Agarose Loading Dye | |||
| 1x MOPS Buffer | |||
| 3% formaldehyde | Sigma-Aldrich | F8775 | Hazardous //// 50-00-0 |
| 50% formamide | Sigma-Aldrich | 47670 | Hazardous //// 75-12-7 |
| Ethidium bromide (EtBr) 30 μg/ml | Sigma-Aldrich | E1510 | Hazardous //// 1239-45-8 |
| Bromophenol blue | Sigma-Aldrich | B0126 | 115-39-9 |
| SMALL RNA FRACTIONATION | |||
| Flat gel loading tips | Life Technologies | LC1002 | |
| Savant SpeedVac Concentrator | Thermo Scientific | DNA120 | |
| Vertical Electrophoresis, Mini-PROTEAN Tetra Cell | Biorad | 165-8000 | |
| Denaturing Acrilamide Gel | |||
| 10% acrylamide/bis-acrylamide (19:1) | Sigma-Aldrich | A3449 | Hazardous. Let the gel polymerizefor 45 min before use |
| 1x TBE Buffer (or 1x MOPS Buffer) | |||
| 7 M urea | Sigma-Aldrich | U6504 | 57-13-6 |
| 0.1% ammonium persulfate | Sigma-Aldrich | 215589 | 7727-54-0 |
| 0.1% TEMED | Sigma-Aldrich | T9281 | 110-18-9 |
| Gene Ruler Ultra Low Range DNA Ladder (10 bp-step) | Thermo Scientific | SM1211 | Label 0.1 μg, as described in the "probe labeling" reaction |
| Acrylamide Blue Dye | |||
| 50% formamide | Sigma-Aldrich | 47670 | Hazardous //// 75-12-7 |
| Bromophenol blue | Sigma-Aldrich | B0126 | 115-39-9 |
| Running Buffer (RB) | |||
| 1x MOPS Buffer | |||
| Alternative RB: 1x TBE Buffer | |||
| 1x TBE Buffer | |||
| 5X TBE | |||
| 445 mM Tris-base | Sigma-Aldrich | T1503 | 77-86-1 |
| 445 mM boric acid | Sigma-Aldrich | B7901 | 10043-35-3 |
| 20 mM EDTA | Sigma-Aldrich | EDS | 60-00-4 |
| Gel Staining Solution | |||
| Ethidium bromide 0.5 μg/ml | Sigma-Aldrich | E1510 | Hazardous //// 1239-45-8 |
| 1x RB | Stain for 15 min with EtBr, destain for 15 min w/o EtBr | ||
| BLOTTING | |||
| 3MM Whatman paper | Sigma-Aldrich | Z270849 | |
| Neutral Nylon Membrane, Hybond NX | GE Healthcare Life Science | RPN203T | Photosensitive |
| CROSSLINKING | |||
| Crosslinking Solution (XLS) | |||
| 1% 1-methylimidazole (v/v) | Sigma-Aldrich | 336092 | Always prepare a fresh aliquot of XLS //// 616-47-7 |
| 12.5 mM HCl | Sigma-Aldrich | H1758 | Hazardous //// 7647-01-0 |
| 3.1% EDC (w/v) | Sigma-Aldrich | 3450 | 25952-53-8 |
| (PRE)-HYBRIDIZATION | |||
| Liquid Scintillation Counter | Beckmann | LS 6500 | |
| Sheared Salmon Sperm, 0.1 mg/ml | Life Technologies | AM9680 | |
| rasi4 antisense probe | 5' CGGUGUUCGACAGUUCCUCGGG -3' | ||
| 5s-rRNA antisense probe | 5'-CAACACGCGGTGTTCCCAAGCCG-3' | ||
| Dmel-miR279 antisense probe | 5'-TTAATGAGTGTGGATCTAGTCA-3' | ||
| Dmel-miR286 antisense probe | 5'-AGCACGAGTGTTCGGTCTAGTCA-3' | ||
| Purification Kit, Illustra MicroSpin G-25 Columns | GE Healthcare Life Science | 27-5325-01 | |
| (Pre)-Hybridization Solution (HS) | |||
| 6x SSPE | Pre-warm HS at 37 ˚C before use | ||
| 0.5% SDS | Sigma-Aldrich | 71736 | 151-21-3 |
| 5x Denhardt's Solution | |||
| 20X SSPE | |||
| 3.6 M NaCl | Sigma-Aldrich | S3014 | 7647-14-5 |
| 0.2 M sodium phosphate | Sigma-Aldrich | 342483 | 7601-54-9 |
| 20 mM EDTA (pH 8.0) | Sigma-Aldrich | EDS | 60-00-4 |
| 50x Denhardt's Solution | |||
| 1% Ficoll 400 (w/v) | Sigma-Aldrich | F2637 | 26873-85-8 |
| 1% Polyvinylpyrrolidone | Sigma-Aldrich | PVP40 | 9003-39-8 |
| 1% BSA | Sigma-Aldrich | A7906 | 9048-46-8 |
| 1X TEN Buffer | |||
| 10 mM Tris-Cl (pH 8.0) | Sigma-Aldrich | T1503 | 77-86-1 |
| 100 mM NaCl | Sigma-Aldrich | S3014 | 7647-14-5 |
| 1 mM EDTA (pH 8.0) | Sigma-Aldrich | EDS | 60-00-4 |
| Probe Labelling | |||
| 0.4 μM oligonucleotide | |||
| 1.5 μl Ci/μl [γ-32P] ATP | Perkin Elmer | NEG002A | Hazardous //// 51963-61-2 |
| 1x T4 Polynucleotide Kinase Buffer | Biolabs | B0201 | |
| T4 Polynucleotide Kinase 0.5 units/μl | Biolabs | M0201 | EC 2.7.1.78 |
| 30 min at 37 ˚C, stop by 20 mM EDTA | |||
| WASHING | |||
| Geiger Mueller Detectors | Canberra Industries | MCB2/CPS – EM77021 | |
| Washing Buffer (WB), 2x SSPE | |||
| Stringent Washing Buffers | |||
| 0.2x SSPE | |||
| 2x SSPE, 0.5% SDS | |||
| 0.2x SSPE, 0.5% SDS | |||
| SIGNAL DETECTION | |||
| PharosFX Plus System | Biorad | 170-9460 | |
| Image J | Freely available | ||
| Quantity One | Biorad | ||
| X-ray films , BioMax XAR | Sigma-Aldrich | F5888 | Photosensitive |
| Developer for X-ray films | Sigma-Aldrich | P7042 | |
| Fixer for X-ray films | Sigma-Aldrich | P7167 | |
| STRIPPING | |||
| Stripping Solution | |||
| 10 mM Tris-HCl pH 8.8 | Sigma-Aldrich | T1503 | 77-86-1 |
| 5 mM EDTA | Sigma-Aldrich | EDS | 60-00-4 |
| 0.1% SDS | Sigma-Aldrich | 71736 | Add SDS after boiling to avoid foaming //// 151-21-3 |
References
- Gesteland, R. F., Cech, T. R., Atkins, J. F. . The RNA World. , (2006).
- Mercer, T. R., Dinger, M. E., Mattick, J. S. Long non-coding RNAs: insights into functions. Nat Rev Genet. 10, 155-159 (2009).
- Salmena, L., Poliseno, L., Tay, Y., Kats, L., Pandolfi, P. P. A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language?. Cell. 146, 353-358 (2011).
- Cesana, M., et al. A long noncoding RNA controls muscle differentiation by functioning as a competing endogenous RNA. Cell. 147, 358-369 (2011).
- Orphanides, G., Reinberg, D. A unified theory of gene expression. Cell. 108, 439-451 (2002).
- Mehler, M. F., Mattick, J. S. Noncoding RNAs and RNA editing in brain development, functional diversification, and neurological disease. Physiol Rev. 87, 799-823 (2007).
- Fire, A., Xu, S., Montgomery, M., Kostas, S., Driver, S., Mello, C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 391, 806-811 (1998).
- Hamilton, A., Baulcombe, D. A species of small antisense RNA in posttranscriptional gene silencing in plants. Science. 286, 950-952 (1999).
- Lee, R. C., Feinbaum, R. L., Ambros, V. The C. Elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 75, (1993).
- Wightman, B., Ha, I., Ruvkun, G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell. 75, 855-862 (1993).
- Fabian, M. R., Sonenberg, N., Filipowicz, W. Regulation of mRNA translation and stability by microRNAs. Annu Rev Biochem. 79, 351-379 (2010).
- Pasquinelli, A. E., et al. Conservation of the sequence and temporal expression of let-7 heterochronic regulatory RNA. Nature. 408, 86-89 (2000).
- Lagos-Quintana, M., Rauhut, R., Lendeckel, W., Tuschl, T. Identification of novel genes coding for small expressed RNAs. Science. 294, 853-858 (2001).
- Lau, N. C., Lim, L. P., Weinstein, E. G., Bartel, D. P. An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science. 294, 858-862 (2001).
- Lee, R. C., Ambros, V. An extensive class of small RNAs in Caenorhabditis elegans. Science. 294, 862-864 (2001).
- Laneve, P., et al. The Gcm/Glide molecular and cellular pathway: new actors and new lineages. Dev Biol. 375, 65-78 (2013).
- Hosoya, T., Takizawa, K., Nitta, K., Hotta, Y. Glial cells missing: a binary switch between neuronal and glial determination in Drosophila. Cell. 82, 1025-1036 (1995).
- Jones, B. W., Fetter, R. D., Tear, G., Goodman, C. S. Glial cells missing: a genetic switch that controls glial versus neuronal fate. Cell. 82, 1013-1023 (1995).
- Vincent, S., Vonesch, J. L., Giangrande, A. Glide directs glial fate commitment and cell fate switch between neurons and glia. Developement. 122, 131-139 (1996).
- Cattenoz, P. B., Giangrande, A. Lineage specification in the fly nervous system and evolutionary implications. Cell Cycle. 12, 2753-2759 (2013).
- Jones, B. W., Abeysekera, M., Galinska, J., Jolicoeur, E. M. Transcriptional control of glial and blood cell development in Drosophila: cis-regulatory elements of glial cells missing. Dev Biol. 266, 374-387 (2004).
- Ragone, G., et al. Transcriptional regulation of glial cell specification. Dev Biol. 255, 138-150 (2003).
- Akiyama-Oda, Y., Hosoya, T., Hotta, Y. Asymmetric cell division of thoracic neuroblast 6-4 to bifurcate glial and neuronal lineage in Drosophila. Development. 126, 1967-1974 (1999).
- Bernardoni, R., Kammerer, M., Vonesch, J. L., Giangrande, A. Gliogenesis depends on glide/gcm through asymmetric division of neuroglioblasts. Dev Biol. 216, 265-275 (1999).
- Ragone, G., Bernardoni, R., Giangrande, A. A novel mode of asymmetric division identifies the fly neuroglioblast 6-4T. Dev Biol. 235, 74-85 (2001).
- Soustelle, L., Roy, N., Ragone, G., Giangrande, A. Control of gcm RNA stability is necessary for proper glial cell fate acquisition. Mol Cell Neurosci. 37, 657-662 (2008).
- Akiyama, Y., Hosoya, T., Poole, A. M., Hotta, Y. The gcm-motif: a novel DNA-binding motif conserved in Drosophila and mammals. Proc. Natl. Acad. Sci. USA. 25, 14912-14916 (1996).
- Miller, A. A., Bernardoni, R., Giangrande, A. Positive autoregulation of the glial promoting factor glide/gcm. EMBO J. 17, 6316-6326 (1998).
- Krichevsky, A. M., King, K. S., Donahue, C. P., Khrapko, K., Kosik, K. S. A microRNA array reveals extensive regulation of microRNAs during brain development. RNA. 9, 1274-1281 (2003).
- Liu, C. G., et al. An oligonucleotide microchip for genome-wide microRNA profiling in human and mouse tissues. Proc. Natl. Acad. Sci. USA. 101, 9740-9744 (2004).
- Yin, J. Q., Zhao, R. C., Morris, K. V. Profiling microRNA expression with microarrays. Trends Biotechnol. 26, 70-76 (2008).
- Schmittgen, T. D., Jiang, J., Liu, Q., Yang, L. A high-throughput method to monitor the expression of microRNA precursors. Nucleic Acids Res. 32, E43 (2004).
- Chen, C., et al. Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res. 33, e179 (2005).
- Shi, R., Chiang, V. L. Facile means for quantifying microRNA expression by real-time PCR. Biotechniques. 39, 519-525 (2005).
- Raymond, C. K., Roberts, B. S., Garrett-Engele, P., Lim, L. P., Johnson, J. M. Simple quantitative primer-extension PCR assay for direct monitoring of microRNAs and short-interfering RNAs. RNA. 11, 1737-1744 (2005).
- Wang, Z., Gerstein, M., Snyder, M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 10, 57-63 (2009).
- Linsen, S. E., et al. Limitations and possibilities of small RNA digital gene expression profiling. Nat Methods. 6, 474-476 (2009).
- Valoczi, A., et al. Sensitive and specific detection of microRNAs by northern blot analysis using LNA-modified oligonucleotide probes. Nucleic Acids Res. 32, e175 (2004).
- Varallyay, E., Burgyan, J., Havelda, Z. Detection of microRNAs by Northern blot analyses using LNA probes. Methods. 43, 140-145 (2007).
- Kim, S. W., et al. A sensitive non-radioactive northern blot method to detect small RNAs. Nucleic Acids Res. 38, e98 (2010).
- Pall, G. S., Hamilton, A. J. Improved northern blot method for enhanced detection of small RNA. Nat Protoc. 3, 1077-1084 (2008).
- Hermanson, G. T. . Bioconjugate Techniques. , (2008).
- Pall, G. S., Codony-Servat, C., Byrne, J., Ritchie, L., Hamilton, A. Carbodiimide-mediated cross-linking of RNA to nylon membranes improves the detection of siRNA, miRNA and piRNA by northern blot. Nucleic Acids Res. 35, e60 (2007).
- Guzzardo, P. M., Muerdter, F., Hannon, G. J. The piRNA pathway in flies: highlights and future directions. Curr Opin Genet Dev. 23, 44-52 (2013).
- Aravin, A. A., et al. Double-stranded RNA-mediated silencing of genomic tandem repeats and transposable elements in the D. melanogaster germline. Curr. Biol. 11, 1017-1027 (2001).
- Vagin, V. V., et al. A distinct small RNA pathway silences selfish genetic elements in the germline. Science. 313, 320-324 (2006).
- Brennecke, J., et al. Discrete small RNA-generating loci as master regulators of transposon activity in Drosophila. Cell. 128, 1089-1103 (2007).
- Sullivan, W., Ashburner, M., Hawley, R. S. . Drosophila protocols. , (2008).
- Zamore, P. D., Ma, S. Isolation of Drosophila melanogaster testes. J. Vis. Exp. (51), e2641 (2011).
- Sambrook, J., Fritsch, E. F., Maniatis, T. . Molecular cloning : a laboratory manual. , (1989).
- Alwine, J. C., Kemp, D. J., Stark, G. R. Method for detection of specific RNAs in agarose gels by transfer to diazobenzyloxymethyl-paper and hybridisation with DNA probes. Proc. Natl. Acad. Sci. USA. 74, 5350-5354 (1977).
- Hansen, T. B., et al. Natural RNA circles function as efficient microRNA sponges. Nature. 495, 384-388 (2013).
- Flores-Jasso, C. F., Salomon, W. E., Zamore, P. D. Rapid and specific purification of Argonaute-small RNA complexes from crude cell lysates. RNA. 19, 271-279 (2013).
- Specchia, V., et al. Hsp90 prevents phenotypic variation by suppressing the mutagenic activity of transposons. Nature. 463, 662-665 (2010).
- Várallyay, E., Burgyán, J., Havelda, Z. MicroRNA detection by northern blotting using locked nucleic acid probes. Nat Protoc. 3, 190-196 (2008).
- Koscianska, E., Starega-Roslan, J., Czubala, K., Krzyzosiak, W. J. High-resolution northern blot for a reliable analysis of microRNAs and their precursors. ScientificWorldJournal. 11, 102-117 (2011).
