

Summary
MicroRNAs (miRNAs) are a widely conserved class of regulatory molecules. Here we describe a miRNA cloning method that relies upon two potent ligation steps followed by high-throughput sequencing. Our method permits accurate genome-wide quantitation of miRNAs.
Abstract
MiRNA cloning and high-throughput sequencing, termed miR-Seq, stands alone as a transcriptome-wide approach to quantify miRNAs with single nucleotide resolution. This technique captures miRNAs by attaching 3’ and 5’ oligonucleotide adapters to miRNA molecules and allows de novo miRNA discovery. Coupling with powerful next-generation sequencing platforms, miR-Seq has been instrumental in the study of miRNA biology. However, significant biases introduced by oligonucleotide ligation steps have prevented miR-Seq from being employed as an accurate quantitation tool. Previous studies demonstrate that biases in current miR-Seq methods often lead to inaccurate miRNA quantification with errors up to 1,000-fold for some miRNAs1,2. To resolve these biases imparted by RNA ligation, we have developed a small RNA ligation method that results in ligation efficiencies of over 95% for both 3’ and 5′ ligation steps. Benchmarking this improved library construction method using equimolar or differentially mixed synthetic miRNAs, consistently yields reads numbers with less than two-fold deviation from the expected value. Furthermore, this high-efficiency miR-Seq method permits accurate genome-wide miRNA profiling from in vivo total RNA samples2.
Introduction
High-throughput sequencing based methodologies have been widely applied to many biological samples in recent years greatly expanding our understanding of the molecular complexity of biological systems3,4. However, preparation of RNA samples for high-throughput sequencing often imparts specific biases inherent to the employed methodology, limiting the potential utility of these powerful techniques. These method specific biases have been well documented for ligation-based, small-RNA library preparations1,2,5,6. These biases result in 1,000-fold variation in reads numbers for equimolar synthetic miRNAs, making inference of miRNA abundance from sequencing data wildly variable and error prone.
Studies focusing on the properties of phage-derived T4 RNA ligases have documented that the enzymes exhibit nucleotide-based preferences7, which manifest as biased libraries in high-throughput sequencing experiments1,2,8. In order to minimize the biases imparted by RNA ligases, multiple strategies have been employed; macromolecular crowding9, randomizing the nucleotide sequence on the adapter which is proximal to the ligation site6, and employing high concentrations of ligation adapter2. Through a combination of these three approaches we have developed a work-flow for unbiased preparation of small RNA libraries compatible for high-throughput sequencing (Figure 1). For direct comparisons between current protocols and our optimized method, please refer to our recent report2. This optimized method yields ligation efficiencies of greater than 95% at both 3’ and 5’ steps and permits the unbiased ligation of small RNA molecules from synthetic and biological samples2.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
NOTE: It is critical to maintain RNase-free conditions during the entire procedure.
1. Adenylation of 3’ Linker
- Dilute DNA oligonucleotide designed for 3’ ligation reactions to 100 μM in nuclease-free H2O.
NOTE: The oligonucleotide should have a 5’ phosphate group, a randomized dinucleotide at the 5’ end, and a 3’ dideoxycytosine. The 5’ phosphate group is a requirement of the Mth enzyme for efficient 5’ adenylation10. The phosphate can be added by pre-treatment of the oligonucleotide with a polynucleotide kinase, or ordered with the modification directly. The randomized dinucleotide at the 5’ end is important for minimizing the sequence preference which RNA ligases exhibit for certain end-end joining reactions over others6. The 3’ dideoxycytosine places a blocking element at the 3’ end of the oligonucleotide to inhibit the formation of linker-linker concatamers during subsequent ligation steps and also serves to block the 3’ end of the miRNA-Linker molecule formed at the end of the 3’ ligation reaction11. The oligonucleotide linker sequence chosen for the unbiased miR-Seq method is as follows: 5’PO4NNTGGAATTCTCGGGTGCCAAGG-dideoxyC3’. - Assemble the following adenylation reaction: 200 pmol 3’ linker oligonucleotide, 4 μl 10x 5’ DNA adenylation buffer, 4 μl 1 mM ATP, 4 μl Mth RNA ligase, nuclease-free H2O to a final volume of 40 μl.
- Incubate reaction at 65 °C for 1 hr. Terminate reaction by heating to 85 °C for 5 min.
- Precipitate adenylated linker by the addition of 2.5 volumes of 100% ethanol and 1/3 volume of 10 M ammonium acetate to remove residual ATP and to prevent unanticipated ligations in future reactions.
- Briefly, mix the alcohol, salt, and oligo solution to homogeneity, spinning at full speed in a microcentrifuge (~15,000 x g) at 4 °C for 20 min. Wash the pellet in 70% ethanol, air dry, and resuspend in the appropriate volume of nuclease-free H2O to yield 50-100 μM of adenylated oligo.
- Confirm adenylation by running an 18% polyacrylamide gel.
- Prepare the gel by mixing the following together: 6.3 g urea, 6.75 ml of 40% acrylamide, and 1.5 ml of 10x TBE. Add H2O to 15 ml.
- Mix until urea is completely dissolved and add 150 µl 10% (w/v) ammonium persulfate (APS) and 15 µl tetramethlyethylenediamine (TEMED).
- Once gel has set, pre-run for 30 min at 24 V/cm of gel width with 1x TBE running buffer at room temperature. Once 30 min is up flush wells with running buffer.
- Mix 5 pmol of adenylated oligonucleotides with and equal volume of 2x denaturing RNA loading buffer (95% (v/v) Formamide, 0.02% (w/v) SDS, 0.02% bromophenol blue (w/v),0.01% (w/v) Xylene Cyanol, 1.0 mM EDTA), heat briefly to 75 °C, and snap cooled on ice. Dispense entire sample into the well of the gel. Also include an identical amount of non-adenylated control, and low molecular weight size standards.
- Run the gel at 24 V/cm until the bromophenol blue is ¾ of the way to the bottom of the gel. Disassemble apparatus, post-stain gel with 1x sybr-gold solution in 1x TBE and visualize on UV-transilluminator box (Figure 2).
NOTE: Gel purify the adenylated oligo in a manner similar to that described above if “no input RNA” controls are resulting in a high amount of PCR product as compared to samples with input RNA.
2. Linker Ligation to 3’ End of miRNA
- 3’ Ligation
- Assemble the following components on ice in the order listed: 1.0 μl 50% (w/v) PEG 8000, 0.5 μl 10x RNA ligase buffer (500 mM Tris-HCl pH 7.5, 100 mM MgCl2, 10 mM DTT), 1.0 μl adenylated 3’ Linker (100 μM), 0.5 μl RNase inhibitor, 0.5-8.0 μg total RNA, 0.5 μl T4 RNA ligase 2, nuclease-free H2O to a final volume of 5 μl.
- Mix the reaction by pipetting as the solution is too viscous to vortex due to the high concentration of PEG 8000. Once thoroughly mixed, place the reaction in a thermal cycler with a heated lid. Set the block to 16 °C, and incubate for 4 hr.
NOTE: The reaction may be scaled up to a final volume of 20 μl without any loss in ligation efficiency. React for 4 hr at ambient temperature or overnight at 4 °C to yield essentially identical ligation efficiencies.
- Gel Purification
- Following 3’ ligation, mix the reaction with an equal volume of 2x RNA loading buffer. Heat to 70 °C for 5 min, and snap cool on ice.
- Run the sample on a 15% polyacrylamide gel for PAGE purification.
- Prepare a 15% polyacrylamide gel containing 7 M urea, in a fashion similar to that described previously in step 1.5.
- Pre-run the gel at 24 V/cm of gel width for at least 30 min. Turn off power supply, flush wells, load sample and run gel at same voltage as pre-run.
- Load the sample and run the PAGE until the bromophenol blue has just exited the gel. The desired product will migrate close to the xylene cyanol.
- Staining, Visualization, and Excision
- Disassemble gel apparatus and place gel in clean tray with 1x running buffer (TBE), 1x sybr-gold, and rock for 15 min.
- Remove gel from staining dish and visualize on UV transilluminator box covered with clean plastic wrap. The ligation product should be around 45 nucleotides in length.
- Excise the region containing the ligation product with a clean razor blade and place in 0.5 ml micro-centrifuge tube.
- Product Isolation and Precipitation
- Pierce bottom of 0.5 ml microcentrifuge tube with a 30 G needle and place the pierced tube in a second clean, low-retention, 1.5 ml microcentrifuge tube. Centrifuge for 5 min at ~15,000 x g.
- Visually confirm that the entire gel slice has moved through the hole in the inner tube. If necessary, use a clean pipette tip to push some gel fragments closer to the hole.
- Discard the empty 0.5 ml tube and add 400 μl of high-salt column buffer (HSCB 400 mM NaCl, 25 mM Tris-HCl pH 7.5, 0.1% SDS) to the acrylamide slurry. Rock tube(s) at 4 °C overnight.
- Transfer slurry to a 0.22 micron cellulose acetate spin column using a wide-bore pipette tip. Centrifuge for 3 min at room temperature at ~15,000 x g to collect all liquid away from acrylamide gel matrix.
- Transfer the liquid to a clean, low-retention, 1.5 ml tube containing 1 μl of 20 mg/ml glycogen. Add an equal volume of isopropanol and 1/10th volume sodium acetate. Mix solution by inverting tube several times and precipitate in -80 °C freezer for 20 min.
- Centrifuge at full speed at 4 °C for 20 min to pellet the nucleic acid. Remove supernatant and wash in 70% ethanol. Air dry pellet for 10 min at room temperature and proceed directly to 5’ ligation step.
3. Linker Ligation to 5’ End of miRNA-3’ Linker Hybrid
- To pelleted nucleic acid, add the following components: 2 µl PEG 8000 (50% w/v), 0.5 µl 10x RNA Ligase Buffer, 0.5 µl ATP (10 mM), 0.5 µl NN-RNA oligo (100 µM), and H2O to a final volume of 4 µl.
NOTE: The composition of the ‘NN-RNA oligo’ is as follows 5’-inverted dideoxy-TCUACAGUCCGACGAUCNN3’ and was purified via HPLC by the manufacturer. - Mix this sample by pipetting up and down repeatedly. Do this for some time (up to 5 min) to ensure that the pellet has been completely re-solubilized. If re-solubilization is problematic, heat the sample briefly (3-5 min) to 37 °C and resume pipetting to mix.
- Once the pellet is back in solution, transfer the contents to a clean PCR tube containing 1 μl of a 1:1 mixture of RNase inhibitor and T4 RNA Ligase 1 (5 units). Again mix by pipetting up and down.
- Incubate the reaction in a thermal cycler at 37 °C for 4 hr. Proceed immediately to cDNA synthesis.
4. Reverse Transcription/cDNA Synthesis
- Add the following components to the 5’ Ligation reaction: 2 µl 5x RT buffer, 0.75 µl 100 mM DTT, 1 µl Illumina RT Primer 5’-GCCTTGGCACCCGAGAATTCCA-3’ (1 µM), and 0.5 µl dNTPs (10 mM).
- Mix by pipetting and heat to 65 °C for 5 min. Then slowly cool on the bench top.
- Once reaction has cooled to room temperature, place tube on ice and add 0.5 μl each of reverse transcriptase enzyme and RNase inhibitor. Mix by pipetting and place in thermal cycler for the extension step indicated in the manufacturer’s instructions.
- Once complete add 5 μl of nuclease-free H2O to bring the final volume of the cDNA library up to 15 μl.
5. Library Preparation
- For the indexing PCR, assemble the following reaction on ice: 6 µl 5x Phusion Buffer, 1 µl dNTPs (10 mM), 0.9 µl DMSO, 0.75 µl 5’ Primer (25 µM), 0.75 µl 3’ Primer (25 µM), 3 µl cDNA library (variable concentration depending on the amount of the input), 0.5 µl Phusion Polymerase (1 unit), nuclease-free H2O to a final volume of 30 μl. Choose the primer sequences to assure compatibility with the template and the sequencing platform of choice.
- Run the PCR reaction with the following settings: initial denaturation at 98 °C for 3 min, for the first 4 cycles, denature 98 °C for 15 sec, anneal at 50 °C for 15 sec, extend at 72 °C for 15 sec.
NOTE: Depending on the amount of the input material, vary the total PCR cycle numbers. Typically, for an input of 1-2 µg of total RNA, 13 total PCR cycles are sufficient to generate the miR-Seq library. - Carry out subsequent cycles, e.g., cycles 5-12, in a two-step manner where the annealing and extension cycles are combined and carried out at 70 °C for 30 sec and the denaturation is again 98 °C for 15 sec.
NOTE: The reaction volume is 30 μl to permit pausing the PCR program and remove a 10 μl aliquot at pre-determined cycle numbers to monitor for appearance of desired product. Typically, cycles 10, 13, and 16 are chosen for testing from a total RNA sample.
- Run the PCR reaction with the following settings: initial denaturation at 98 °C for 3 min, for the first 4 cycles, denature 98 °C for 15 sec, anneal at 50 °C for 15 sec, extend at 72 °C for 15 sec.
- Prepare an 8% non-denaturing acrylamide gel by mixing 2 ml of 40% acrylamide, 1 ml of 10x TBE, and H2O to 10 ml. Then add 100 μl of 10% APS and 10 μl of TEMED. Pour gel and let set.
- Pre-run the gel at 12 V/cm of gel width for 30 min. Flush wells. Add 1 μl of 10x loading buffer (15% Ficoll-400, 66 mM EDTA, 20 mM Tris-HCl pH 8.0, 0.1% (w/v) SDS, 0.01% (w/v) bromophenol blue), add 10 μl of sample to each well of dimensions 5 mm x 1 mm x 15 mm. Typically, each well can load up to 30 μl of samples. Run the sample gel at 10 V/cm of gel width in 1x TBE buffer until the bromophenol blue dye just runs out of the bottom of the gel.
- Stain and visualize gel as described in step 2.3.
- Excise the 146 base pair band and elute overnight in 0.25 ml of HSCB in a fashion similar to what is described in section 2.4. Precipitate DNA in an equal volume of isopropanol and 1/10th volume sodium acetate. Wash pellet in 70% ethanol and resuspend in TE buffer.
- Quantify the library using picogreen according to manufacturer’s recommendations.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
The anticipated results for the preceding method should initially be observation of a single nucleotide shift (increase) in the size of the DNA oligonucleotide that was subject to adenylation by Mth RNA ligase (Figure 2). Following 3’ ligation, visualization of the acrylamide gel indicates (see Figure 3) sharp high molecular weight bands evident in the 100-300 nucleotide region of the gel. This indicates that the total RNA sample being used is of high quality (not degraded). Secondly one should observe a very bright signal at the 25 nucleotide region of the gel, which is excess, unligated 3’ linker. It also may be helpful to include a no input RNA control in the 3’ ligation. This will indicate the purity of the 3’ linker, and also can be carried through to the PCR step for indication of the cycle number when non-specific signal becomes problematic. There is no diagnostic for the 5’ ligation, however shown in Figure 4 is a reaction identical to the one described carried out with radio-labeled RNA which indicates the importance of the high concentration of PEG8000 employed. Finally, after the PCR and native PAGE, a DNA product of 146 base pairs consistent with the size of the adapter sequences, a miRNA insert, and additional sequence from the extended PCR primers can be observed. It is important to note that if the proper number of PCR cycles (which is determined empirically) is exceeded, the no miRNA insert product may obscure the desired amplicon size. Shown in Figure 5 is a result from 5 pmoles of synthetic miRNA, typically for 2 μg of total RNA 13-18 cycles of PCR are necessary.

Figure 1. Schematic of the workflow for 2-step ligation small-RNA library preparation. Shown are general steps for preparation of an unbiased miR-Seq library. Shown in black is 3’ DNA ligation adapter, which is activated via adenylation in step 1, miRNA is shown in green and is ligated to the 3’ adapter in step 2, and 5’ RNA ligation adapter is in blue which is ligated to the chimeric molecule in step 3. Numbered steps are described in detail in the Protocol section, italics indicate enzymatic steps.

Figure 2. Adenylation of 3’ Linker with Mth RNA-Ligase. 18% PAGE-Urea gel from adenylation of DNA oligonucleotide to be used for subsequent 3’ ligation reaction, (+) indicates sample that was incubated with Mth RNA ligase, (-) indicates sample incubated without enzyme, and (m) indicates the size standards (numbers shown at right in base pairs). At left is a schematic of the oligonucleotide species present. Asterisk indicates larger DNA products generated by aberrant oligonucleotide synthesis.

Figure 3. 3’ Ligation of Synthetic and Total RNA Samples. A) 15% PAGE-Urea gel of 3’ ligation reactions, (m) indicates size standards (numbers at left indicate base pairs), (No RNA) indicates a reaction with no input RNA and 3’ linker that was not gel-purified, (+) indicates ligation performed with 5 pmol of synthetic miRNA and a 3’ linker which was gel-purified, (2 and 1 μg) indicate the amount of mouse total RNA subjected to 3’ ligation with the same, gel-purified linker, asterisk indicates excess 3’ linker. B) Autoradiogram of similar 3’ ligation reaction performed with P32 5’ end labeled synthetic miRNA. Number at top indicates time (hr) the reaction was allowed to proceed, and at bottom indicates the amount ligated as a percentage of the sum of unligated and ligated.

Figure 4. 5’ Ligation of Radio-labeled miRNA-3’Linker Hybrid. Autoradiogram of 5’ ligation reaction performed with P32 5’ end labeled synthetic miRNA-3’ linker hybrid. Number at top indicates amount of PEG used in the reaction, and number at bottom indicates the amount ligated as a percentage of the sum of unligated and ligated. Lines at left are a schematic of the ligated molecules where miRNA is in green, 5’ and 3’ adapters are blue and black, respectively.
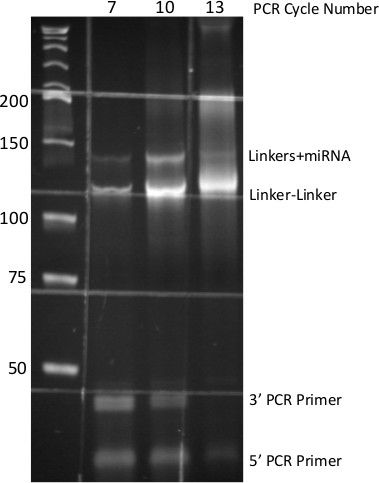
Figure 5. PCR-derived small-RNA DNA Library. 8% Native PAGE of PCR products generated from miR-Seq ligation procedure. Numbers at top indicate cycles of PCR, numbers at side indicate molecular size standards. Each DNA species from the PCR is identified at right. Grid pattern is due to auto-fluorescence of specimen dish.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
The methodology described herein makes use of several key variables to maximize ligation efficiencies, namely high concentrations of PEG, use of randomized linkers, and high concentration of linkers2,6,9. This approach permits reliably quantitative sequencing libraries from total RNA samples2. We have conducted multiple titrations of input RNA and have concluded that the preceding methodology is best suited for total RNA amounts in the 1-8 μg range (data not shown). When amounts in the 10-500 ng range are used, a majority of the read space is consumed by adapter concatamers and bacterial sequences from which the RNA ligases are purified. However, it is worth noting in these low input experiments that the miR species present, though low in reads number, are still reflective of actual amounts when compared to identical higher input samples. In order to ensure that the miR profiles observed from total RNA samples are reflective of actual amounts, and to allow cross-examination of different samples, we routinely include 10-fold dilutions of three synthetic calibrator RNAs12. We have found that the inclusion of 0.1:0.01:0.001 pmol of distinct calibrator RNAs yield useful reads numbers for confirming the quantitative nature of the assay.
As noted in Figure 1 (see asterisk) synthetic oligonucleotides frequently contain aberrant molecules which are distinct in size from the sequence ordered. If these products are able to co-purify with the miRNA-3’ linker hybrid, then it may be necessary to PAGE purify the 3’ linker following Mth adenylation. We have found this to be helpful for reducing the amount of non-specific PCR product observed in negative control samples.
Although the methodology described above was benchmarked for quantitative preparation of miRNA libraries2, we fully anticipate that it is readily applicable to more general procedures as well; namely RNA-seq and CLIP-Seq (Cross Link Immuno-Precipitation) library preparation. In fact, we have employed some of the same principles of the preceding methodology to CLIP-Seq experiments resulting in robust library PCR relative to previously published reports13 (data not shown). Finally, as serum miRNAs gain traction as a medical diagnostic tool, we anticipate the preceding methodology will aid greatly in identification of the most robust and reliable miRNA biomarkers.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
The authors declare no competing financial interests.
Acknowledgments
The authors would like to thank members of the Yi laboratory especially Zhaojie Zhang for fruitful discussions regarding linker design and ligation efficiencies, as well as the American Cancer Society for supporting this work through a postdoctoral fellowship (#125209) to J.E.L. Research reported in this publication was also supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health under Award Number R01AR059697 (to R.Y.) and a research grant from the Linda Crnic Institute for Down Syndrome. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Materials
| Name | Company | Catalog Number | Comments |
| 3' Linker (5' phosphorylated, 3' blocked) | Integrated DNA Technologies | custom | |
| 5' Linker | Integrated DNA Technologies | custom | 5' blocked, HPLC purified |
| T4RNL2 (1-249 K227Q) | New England Biolabs | M0351S | Specialized for ligation of pre-adenylated DNA adapters |
| 10x Ligation buffer (without ATP) | New England Biolabs | Included with M0351S | |
| 10x Ligation buffer (with ATP) | New England Biolabs | Included with M0204L | |
| RNaseOUT | Invitrogen | 10777-019 | |
| Polyethylene glycol (mol. Wt. 8,000) | New England Biolabs | Included with M0204L | |
| Nuclease-free water | Ambion | AM9937 | We have found water collected from a distillation apparatus to be of equvalent quality. |
| T4RNL1 | New England Biolabs | M0204L | |
| Superscript III RT kit | Invitrogen | 18080-051 | |
| Phusion PCR kit | New England Biolabs | M0530S | |
| Illumina RP1 primer | Integrated DNA Technologies | custom | Sequence information available from Illumina |
| Illumina RT primer | Integrated DNA Technologies | custom | Sequence information available from Illumina |
| Illumina index primer(s) | Integrated DNA Technologies | custom | Sequence information available from Illumina |
| 40% Acrylamide | Fisher Scientific | BP14081 | |
| Urea | Sigma Aldrich | U6504 | |
| Ammonium persulfate | Sigma Aldrich | A3678 | |
| Tetramethyethylenediamine (TEMED) | Sigma Aldrich | T9281 | |
| 2x Denaturing RNA loading buffer | New England Biolabs | Included with M0351S | |
| Razor blades | VWR | 55411-050 | |
| SpinX Centricon tubes | Costar | CLS8161 | |
| Low retention microfuge tubes | Fisher Scientific | 02-681-320 | |
| Sybr Gold | Invitrogen | S-11494 | |
| Adenylation kit | New England Biolabs | E2610L |
References
- Hafner, M., et al. RNA-ligase-dependent biases in miRNA representation in deep-sequenced small RNA cDNA libraries. RNA. 17 (9), 1697-1712 (2011).
- Zhang, Z., Lee, J. E., Riemondy, K., Anderson, E. M., Yi, R. High-efficiency RNA cloning enables accurate quantification of miRNA expression by deep sequencing. Genome Biology. 14 (10), 109 (2013).
- Consortium, T. E. P. The ENCODE (ENCyclopedia Of DNA Elements) Project. Science. 306 (5696), 636-640 (2004).
- Consortium, T. E. P. An integrated encyclopedia of DNA elements in the human genome. Nature. 489 (7414), 57-74 (2012).
- Linsen, S. E. V., et al. Limitations and possibilities of small RNA digital gene expression profiling. Nature Methods. 6 (7), 474-476 (2009).
- Jayaprakash, A. D., Jabado, O., Brown, B. D., Sachidanandam, R. Identification and remediation of biases in the activity of RNA ligases in small-RNA deep sequencing. Nucleic Acids Research. 39 (21), 141 (2011).
- McLaughlin, L. W., Romaniuk, E., Romaniuk, P. J., Neilson, T. The Effect of Acceptor Oligoribonucleotide Sequence on the T4 RNA Ligase Reaction. European Journal of Biochemistry. 125 (3), 639-643 (1982).
- Zhuang, F., Fuchs, R. T., Sun, Z., Zheng, Y., Robb, G. B. Structural bias in T4 RNA ligase-mediated 3′-adapter ligation. Nucleic Acids Research. 40 (7), 54 (2012).
- Harrison, B., Zimmerman, S. B. Polymer-stimulated ligation: enhanced ligation of oligo- and polynucleotides by T4 RNA ligase in polymer solutions. Nucleic Acids Research. 12 (21), 8235-8251 (1984).
- Torchia, C., Takagi, Y., Ho, C. K. Archaeal RNA ligase is a homodimeric protein that catalyzes intramolecular ligation of single-stranded RNA and DNA. Nucleic Acids Research. 36 (19), 6218-6227 (2008).
- Lau, N. C., Lim, L. P., Weinstein, E. G., Bartel, D. P. An Abundant Class of Tiny RNAs with Probable Regulatory Roles in Caenorhabditis elegans. Science. 294 (5543), 858-862 (2001).
- Yi, R., et al. DGCR8-dependent microRNA biogenesis is essential for skin development. Proceedings of the National Academy of Sciences. 106 (2), 498-502 (2009).
- Leung, A. K. L., et al. Genome-wide identification of Ago2 binding sites from mouse embryonic stem cells with and without mature microRNAs. Nature Structural & Molecular Biology. 18 (2), 237-244 (2011).
