A radiolabeled IRE probe was prepared, as described in sections 3 and 4 of the protocol. The sequence of the probe was 5'-GGGCGAAUUC GAGCUCGGUA CCCGGGGAUC CUGCUUCAAC AGUGCUUGGA CGGAUCCU-3'; the bolded nucleotides represent an unpaired C residue and the loop, which are critical IRE features. The specific radioactivity of the probe was 4.5 x 109 cpm/μg of RNA.
To assess the effects of iron perturbations on IRE-binding activity, murine RAW264.7 macrophages were left untreated, or treated with hemin (iron source) or desferrioxamine (iron chelator). Lysates were prepared and analyzed by EMSA with the IRE probe (8,000 cpm/μg protein). Representative data are shown in Figure 1, with shorter and longer exposures of the autoradiograms on the left and right panels, respectively. Murine IRE/IRP1 and IRE/IRP2 complexes exhibit different mobility and two distinct bands are visible in lane 1, corresponding to untreated cells. Iron chelation profoundly induced the IRE-binding activities of both IRP1 and IRP2 (lane 2), while iron supplementation diminished them (lane 3). Pretreatment with 2% 2-ME fully activated IRP1 (bottom panel), even in extracts of iron-treated cells. This demonstrates that iron perturbations do not impinge on the stability of IRP1 and also serves as a loading control. By contrast the 2-ME pretreatment inhibited the IRE-binding activity of IRP2. The fast migrating non-specific bands are not affected by iron.
Next, we analyzed IRE-binding activity in livers of wild type, Irp1-/- and Irp2-/- mice, previously fed for one week with a standard or an iron-enriched diet (Figure 2). The Irp1-/- and Irp2-/- mice were kindly provided by Dr. M. W. Hentze (EMBL, Heidelberg). In wild type liver extracts, IRP1 accounted for the major fraction of IRE-binding activity and IRE/IRP2 complexes were hardly visible (lanes 1, 2), in agreement with previous observations26. IRP2 exhibited potent IRE-binding activity in liver extracts of Irp1-/- mice (lanes 3, 4). Under these conditions, no IRE/IRP1 complexes were observed. Likewise, no IRE/IRP2 complexes were formed in liver extracts of Irp2-/- mice (lanes 5, 6). Feeding mice with a high-iron diet decreased hepatic IRE-binding activity of both IRP1 and IRP2; this effect was more dramatic on IRP2 (lanes 7-12). Note that after treatment of wild type or Irp2-/- liver extracts with 2-ME, the IRE-binding activity of IRP1 was enhanced to a point where it shifted almost all IRE probe (this is clearly observed in the shorter exposure of the autoradiograms on the left panel).
Finally, we evaluated the IRE-binding activity in livers and spleens of wild type and Hjv-/- mice (Figure 3). The Hjv-/- mice were kindly provided by Dr. N. C. Andrews (Duke University, NC). These animals represent a model of hereditary hemochromatosis27, a disease of systemic iron overload, where excessive iron accumulates in parenchymal cells, while reticuloendothelial macrophages remain iron deficient28. As expected, livers of Hjv-/- mice (with iron-loaded hepatocytes) exhibited decreased IRE-binding activity compared to wild type (lanes 1-4). Conversely, IRE-binding activity was higher in spleens of Hjv-/- mice (containing iron-deficient macrophages; lanes 5-8). Again here, the 2-ME treatment promoted almost complete shift of the IRE probe in liver extracts (see shorter exposure of the autoradiograms on the left panel).
Table 1. Cytoplasmic lysis buffer.
| 1% Triton X100 |
| 25 mM Tris-HCl, pH 7.4 |
| 40 mM KCl |
- The solution should be autoclaved and stored at RT.
- Protease inhibitors, such as 10 μg/ml leupeptin and 0.1 mM phenylmethanesulfonyl fluoride (PMSF) may be added prior to use.
- Addition of fresh 1 mM dithiotheritol is recomended for detection of IRP2 activity.
Table 2. Stock solutions for in vitro transcription reaction.
| 1 μg/μl linearized plasmid template |
| 5x transcription buffer (supplied with T7 RNA polymerase) |
| 20 mM mix of ATP, CTP and GTP |
| 3,000 Ci/mmol [α-32P]-UTP |
| 100 mM dithiothreitol |
| 10 U/μl RNase inhibitor |
| 20 U/μl T7 RNA polymerase |
Table 3. Stock solutions for non-denaturing polyacrylamide gel electrophoresis.
| 40% acrylamide:bisacrylamide (37.5:1) |
| 5x TBE (Tris/borate/EDTA) |
For making 1 L of 5x TBE, use 54 g of Tris base, 27.5 g of boric acid, and 20 ml of 0.5M EDTA (pH 8.0).
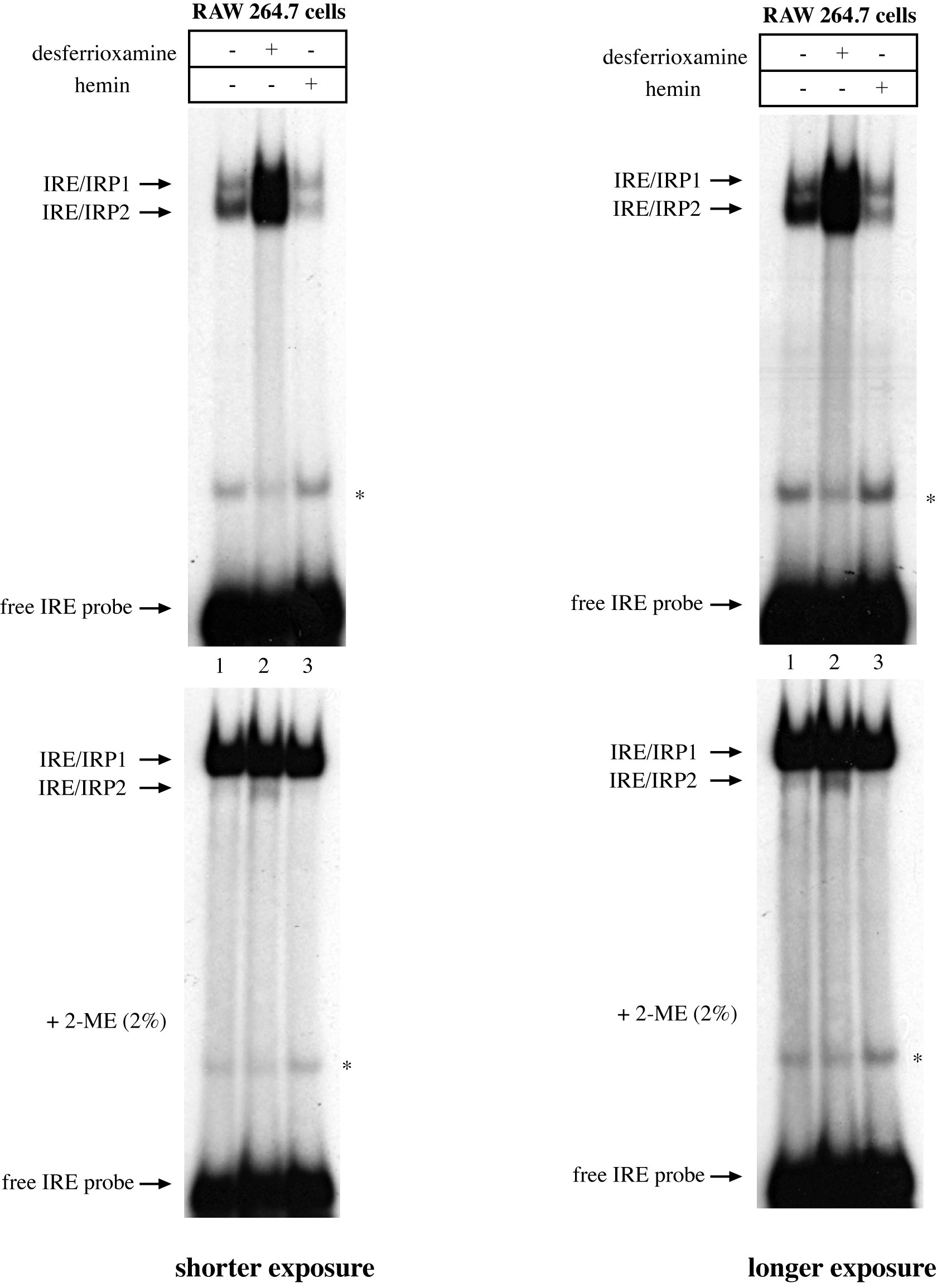
Figure 1. Iron-dependent regulation of IRE-binding activities in RAW264.7 macrophages. 107 cells were either left untreated or treated O/N with 100 μM hemin or desferrioxamine. Cytoplasmic lysates were prepared and analyzed by EMSA with a 32P-labeled IRE probe in the absence (top) or presence of 2% 2-ME (bottom). The positions of IRE/IRP1 and IRE/IRP2 complexes, and of free IRE probe are indicated by arrows. The star indicates a non-specific band. Shorter and longer exposures of the autoradiograms are shown in the left and right panels, respectively. Please click here to view a larger version of this figure.
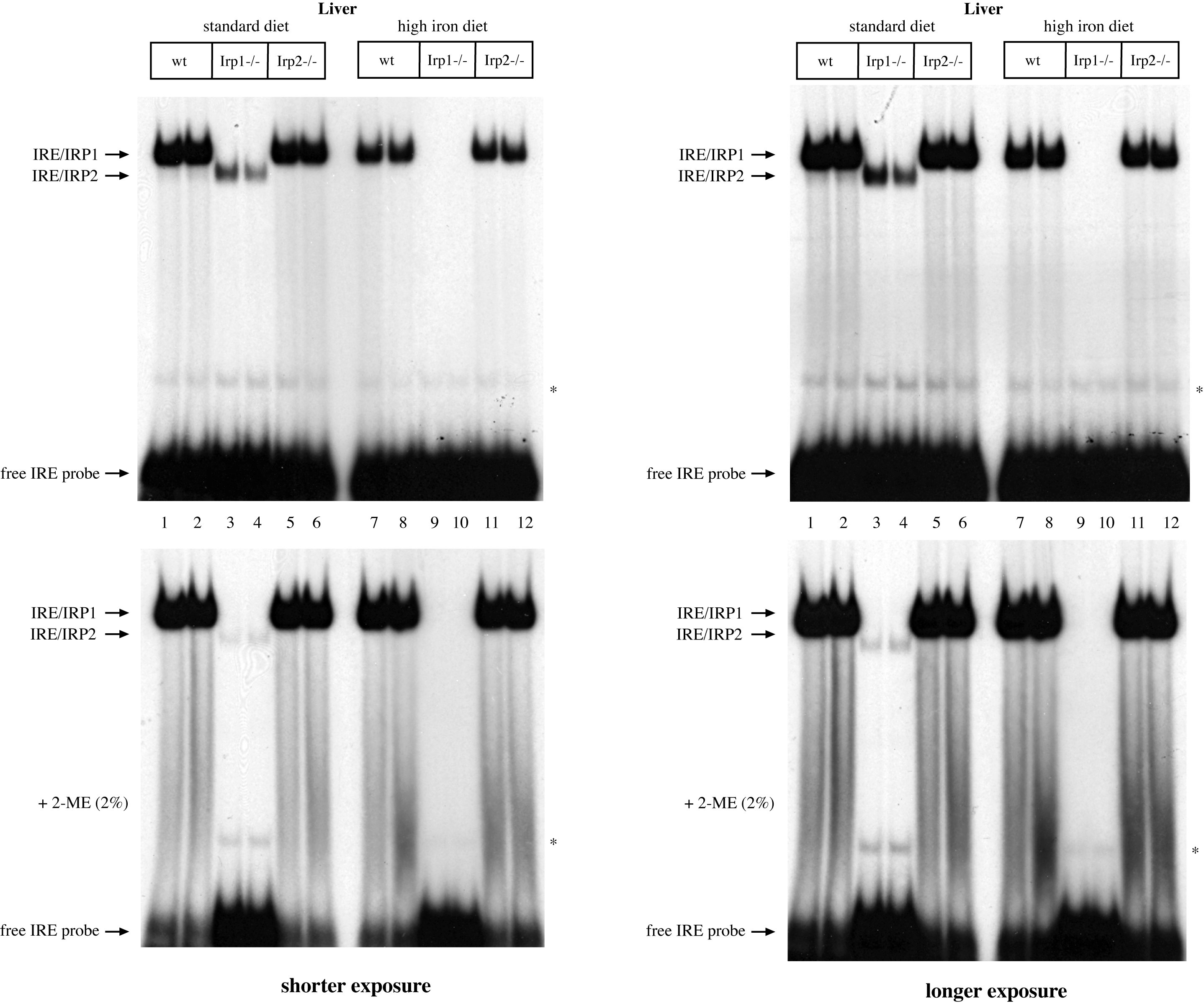
Figure 2. Analysis of IRE-binding activities in livers of wild type (wt), Irp1-/- and Irp2-/- mice. 5 week old mice (n = 2 for each genotype), all in C57BL/6 background29, were placed on a standard or a high-iron diet (containing 2% carbonyl iron). After one week the animals were euthanized. Hepatic protein extracts were prepared and analyzed by EMSA with a 32P-labeled IRE probe in the absence (top) or presence of 2% 2-ME (bottom). The positions of IRE/IRP1 and IRE/IRP2 complexes, and of free IRE probe are indicated by arrows. The star indicates a non-specific band. Shorter and longer exposures of the autoradiograms are shown in the left and right panels, respectively. Please click here to view a larger version of this figure.

Figure 3. Analysis of IRE-binding activities in livers and spleens of wild type (wt) and Hjv-/- mice. 10 week old mice (n = 2 for each genotype), all in C57BL/6 background30, were euthanized. Hepatic and splenic protein extracts were prepared and analyzed by EMSA with a 32P-labeled IRE probe in the absence (top) or presence of 2% 2-ME (bottom). The positions of IRE/IRP1 and IRE/IRP2 complexes, and of free IRE probe are indicated by arrows. The star indicates a non-specific band. Shorter and longer exposures of the autoradiograms are shown in the left and right panels, respectively. Please click here to view a larger version of this figure.
