Multiplexing fluorescent genetically barcoded cells for the purpose of biological applications can only be achieved once individual clonal populations have been generated. Multiplexing is most effective when barcoded populations have clear distinct fluorescent characteristics with minimal spectral overlap. The example shown in Figure 1 with clonal populations of mammalian SupT1 cells illustrates that barcoded cells with mCherry and cyano fluorescent protein (CFP) can be easily analyzed simultaneously without losing their individual fluorescent characteristics. This matrix thus exemplifies a panel of fluorescent genetically barcoded cells that is usable for multiplexing biological applications with a readout in yet an additional available channel. In order to obtain such a panel it is important to remember the nature of retroviral technology, which will lead to variable ranges of fluorescent intensities within the population due to insertional effects and/or MOI. Figure 2 illustrates that initial transduction of mammalian cells with viral particles containing either td Tomato or E2 Crimson results in cells that express either one or both of the fluorescent proteins at a wide range of intensities (left panel). Selection and sorting of single cells into 96-well plates as shown by the gated boxes (mid panel), allows one to obtain tight clonal populations following expansion (right panel). A tight population should be defined as having a small CV which may range according to cell type, normally 30-40% in mean fluorescence intensity. Figure 2 also illustrates that to further increase the number of genetically barcoded populations with a matrix of only two fluorescent proteins, one can also exploit fluorescence intensity. Generally, one log deviation in the mean fluorescence intensity between populations is suitable for achieving appropriate separation from each other following sorting and amplification. Td Tomato was chosen in the shown example to illustrate this feature, where two populations of differing intensities, mid and high, were obtained for multiplexing with E2 Crimson at a single intensity.
Multiplexing is a powerful tool to facilitate the analysis of many samples at the same time and for the ability to decode masked populations. Enhancing multiplexing can be achieved with yet a third fluorescent protein such as eGFP, as long as spectral properties do not interfere with each other. In the experimental procedure illustrated in Figure 3 the panel of six populations obtained with td Tomato and E2 Crimson was exploited to increase multiplexing with eGFP. Retroviral technology was used to transfer eGFP to the populations represented in one of the matrices. When observed in the eGFP channel, the non-green naïve six-population matrix (upper left panel in Figure 3) is indistinguishable from the eGFP-transduced population matrix (lower left panel in Figure 3). Importantly, the panels can be analyzed in the channels occupied by the original genetic barcode (td Tomato and E2 Crimson). While indistinguishable in these channels (compare populations 1-6 with populations 7-12 in mid panels, Figure 3), the individual populations can be analyzed in the eGFP channel, and decoded or tracked back, as shown in the histograms (right panels in Figure 3). After repeating the process of transduction, selection, sorting, and amplification, taking advantage of the unoccupied channels is useless if right compensation is not applied to adjust for possible spectral overlap. To prove this, when populations 1, 2, 3 (non-green) and 7, 8, 9 (green) are analyzed in the eGFP and td Tomato channels, populations 7 and 8 are difficult to distinguish (left panel in Figure 4). It is thus necessary to choose naïve cells (population 1) as a negative control so that the parameters of the instrumentation can be set. When analyzing any matrix, especially with fluorophores that spectrally overlap, it is imperative that single color controls are used to determine the correct compensation values. In the analysis of Figure 4 populations 2 or 3, and 7 serve this purpose, allowing to better define populations that are truly double positive (populations 8 and 9).
Genetically barcoded cells with fluorescent markers retain their own identity as defined by their biosignature, when analyzed in the right fluorescent channel with the right instrument. However, the biosignature becomes just an additional individual property unless exploited for biological applications. In order to prove the power of fluorescent genetic barcoding for multiplexing we decided to introduce one of our previously developed assays for drug discovery into some of the fluorescent barcoded mammalian cell lines. By doing so, fluorescent genetic barcoding was exploited to achieve three assays in one sample. In this procedure a scaffold protein containing one of three putative viral substrate for proteolysis was transferred into the genetically barcoded cells. In the assay, cleavage is revealed by the loss of the FLAG epitope on the cell surface (Figure 5B). In contrast, FLAG surface staining represents lack of cleavage. Figure 5 illustrates the result of the analysis of three different substrates; HIV-1 Envelope wild type (Env wt), HIV-1 Envelope mutant (Env mut), and Dengue Virus (Denv) prM. Each of them was introduced into 1 of 3 barcoded cell lines, naïve, and 2 others utilizing td Tomato at different intensities (mid and high; Figure 5A). Staining for FLAG surface expression reveals which of the substrates was cleaved based on the respective barcode. In the example only HIV Env mut retains the FLAG tag (positive following staining), as seen in the green-FITC channel (Figure 5C, right bottom panel). The analysis of the assay can thus be performed independently of the original barcoding, and be further exploited to decode and track back the distinct populations. This method of multiplexing thus relies on an additional channel reserved for the biological readout of interest.

Figure 1: Barcoding for multiplexing. Individual populations genetically barcoded using distinct fluorescent proteins such as mCherry, CFP or both (left panel) can be mixed, analyzed, and decoded (right panel) in their respective channels via flow cytometry. Adapted from Smurthwaite, C. et al., 201458. Please click here to view a larger version of this figure.

Figure 2: Sorting and amplification of barcoded cells. Mammalian cells were analyzed 48 hr following transduction with viral particles containing either td Tomato or E2-Crimson (left panel). Gates are then set for sorting to include cells expressing td Tomato at different intensities, with/out E2-Crimson (middle panel). Clones from the sorts were amplified and re-analyzed to generate a matrix of six distinguishable populations (right panel). Please click here to view a larger version of this figure.
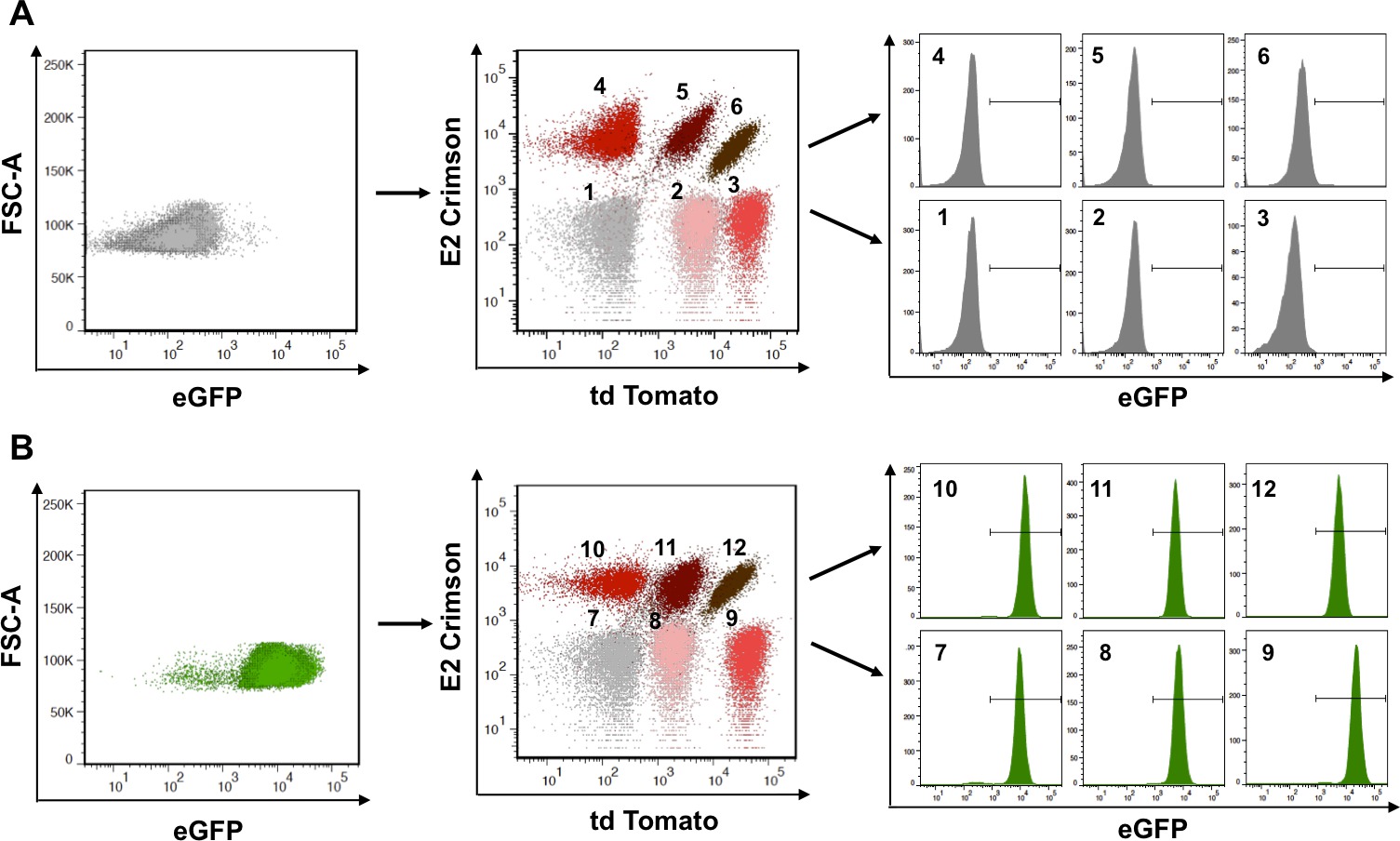
Figure 3: Decoding reveals masked populations. The matrix of six populations obtained in Figure 2 (td Tomato and/or E2-Crimson) can be further engineered to express an additional fluorescent protein such as eGFP. (A) The original matrix, when analyzed in the eGFP channel (left panel) is negative and the six populations are indistinguishable from each other. Each of the six populations can be independently analyzed in the eGFP channel as shown in the histograms (right panels). (B) The same matrix, now expressing also eGFP, was analyzed as in A, revealing now their green fluorescent characteristic. Adapted from Smurthwaite, C. et al., 201458. Please click here to view a larger version of this figure.

Figure 4: Compensation ensures correct separation. Some of the populations originally chosen based on td Tomato and/or eGFP expression (populations 7 and 8) cannot be properly separated when analyzed together (left panel) unless appropriate compensation for these channels is adjusted (right panel). Please click here to view a larger version of this figure.
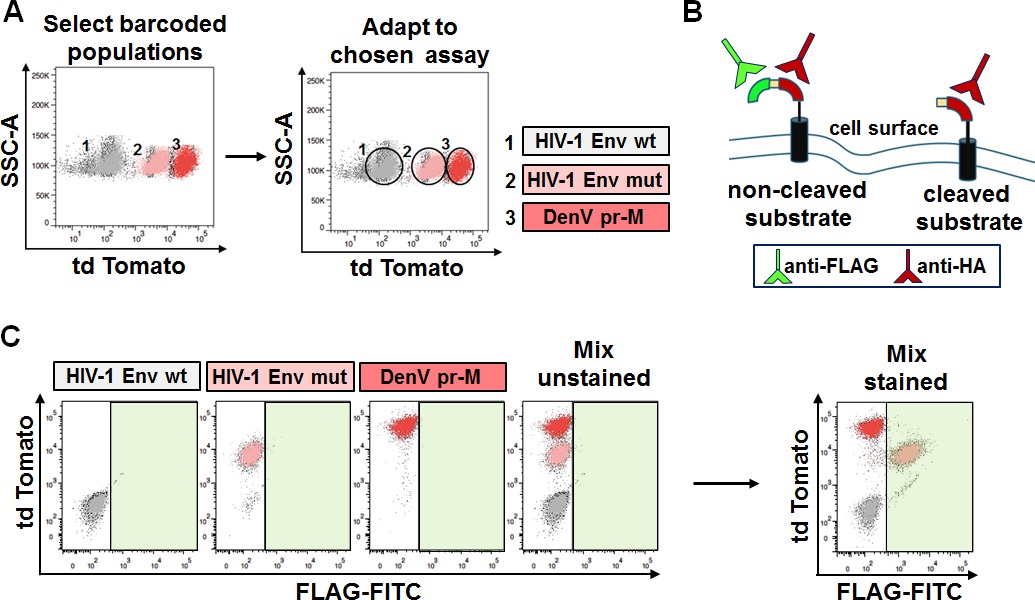
Figure 5: Selection of barcoded cells for further adaptation to chosen assay. (A) Selection and adaptation to assay of choice. Populations genetically barcoded with td Tomato at different intensities (top left panel) were used for biological applications. Each of the populations was further engineered to contain an assay that monitors cleavage, but each one of a different substrate (top right panel). (B) Depiction of the assay. Positive FLAG stain indicates lack of cleavage while negative FLAG stain indicates cleavage. (C) Analysis of the assay following FLAG staining. Mixed populations are distinguishable based on the td Tomato barcode but indistinguishable in the FITC channel (left panels). When stained with FLAG-FITC antibody, only one population is positive for FLAG. Decoding reveals, based on genetic barcoding, that this population bears the HIV Env mut substrate (right panel). Please click here to view a larger version of this figure.
