

Summary
Jämförelsen och optimeringen av två organellära DNA-berikningsmetoder presenteras: traditionell differentiell centrifugering och fraktionering av det totala gDNA baserat på metyleringsstatus. Vi bedömer den resulterande DNA-kvantiteten och kvaliteten, demonstrerar prestanda vid kortläsning av nästa generationens sekvensering och diskuterar potentialen för användning vid långlästsekvens med en molekylsekvensering.
Abstract
Plantorganella genomer innehåller stora, repetitiva element som kan genomgå parning eller rekombination för att bilda komplexa strukturer och / eller subgenomiska fragment. Organellärgener finns också i tillsatser inom en given cell eller vävnadstyp (heteroplasm), och en överflöd av subtyper kan förändras under utveckling eller vid stress (substökiometrisk växling). Nästa generationens sequencing (NGS) -teknologi krävs för att få en djupare förståelse av organellär genomstruktur och funktion. Traditionella sekvenseringsstudier använder flera metoder för att erhålla organellärt DNA: (1) Om en stor mängd startvävnad används, homogeniseras den och utsätts för differentiell centrifugering och / eller gradientrening. (2) Om en mindre mängd vävnad används ( dvs. om frön, materialet eller utrymmet är begränsat) utförs samma process som i (1), följt av hel-genom-amplifiering för att erhålla tillräckligt med DNA. (3) Bioinformatikanalys kan användas för att sökaDet totala genomiska DNA: n och att analysera organellärläsning. Alla dessa metoder har inneboende utmaningar och avvägningar. I (1) kan det vara svårt att erhålla en så stor mängd startvävnad; I (2) kan helgenomförstärkning införa en sekvenseringsförspänning; Och i (3) kan homologi mellan kärn- och organellära genomer störa sammansättning och analys. I växter med stora kärnämnen är det fördelaktigt att berika för organellär DNA för att minska sekvenseringskostnader och sekvenskomplexitet för bioinformatikanalyser. Här jämför vi en traditionell differentialcentrifugeringsmetod med en fjärde metod, en anpassad CpG-metyl-pulldown-tillvägagångssätt, för att separera det totala genomiska DNA-värdet i kärn- och organellära fraktioner. Båda metoderna ger tillräckligt med DNA för NGS, DNA som är högt berikat för organellära sekvenser, om än i olika förhållanden i mitokondrier och kloroplaster. Vi presenterar optimeringen av dessa metoder för vävnadslövvävnad och diskuterar stora fördelar och dNackdelar med varje tillvägagångssätt i samband med provingång, protokolllättnad och nedströmsapplikation.
Introduction
Genomsekvensering är ett kraftfullt verktyg för att dissekera den underliggande genetiska grunden för viktiga växtegenskaper. De flesta genom-sekvensstudierna fokuserar på kärngenominnehållet, eftersom majoriteten av gener ligger i kärnan. Organella genomer, inklusive mitokondrier (över eukaryoter) och plastider (i växter, den specialiserade formen, kloroplasten, arbetar med fotosyntes) bidrar emellertid med väsentlig genetisk information som är väsentlig för organismutveckling, stressrespons och övergripande fitness 1 . Organellärgener ingår typiskt i totala DNA-extraktioner avsedda för kärngenom-sekvensering, även om metoder för att reducera organelltal före DNA-extraktion också används 2 . Många studier har använt sekvenseringsresultat från totala gDNA-extraktioner för att samla organellära genom 3 , 4 , 5 ,Xref "> 6 , 7. Men när målet för studien är att fokusera på organella genomer, ökar sekvenseringskostnaderna eftersom många läsningar är" förlorade "till kärn-DNA-sekvenserna, speciellt i växter med stora kärnvätskor På grund av dubbelarbete och överföring av organella sekvenser i kärngenomet och mellan organeller, är det dessutom en lösning av den korrekta mapppositionen för sekvensering som läser till det ordnade genomet bioinformatiskt utmanande 2 , 8. Rening av organella genomerna från kärngenometet är en Strategi för att minska dessa problem. Ytterligare bioinformatikstrategier kan användas för att separera läser den kartan till regioner med homologi mellan mitokondrier och kloroplaster.
Medan organellärgenerna från många växtarter har sekvenserats, är lite känt om bredden av organellär genom diversitetTillgänglig i vilda populationer eller odlade avelspooler. Organellärgener är också kända för att vara dynamiska molekyler som genomgår signifikant strukturell omläggning på grund av rekombination mellan repetitionssekvenserna 9 . Dessutom finns flera kopior av organellärgenomet inom varje organell, och flera organeller finns inom varje cell. Inte alla kopior av dessa genomer är identiska, vilket är känt som heteroplasmi. I motsats till den kanoniska bilden av "mästarkretsar" finns det nu växande bevis för en mer komplex bild av organellära genomstrukturer, inklusive sub-genomiska cirklar, linjära kromosomer, linjära concatamerar och grenade strukturer 10 . Sammansättningen av växtorganella genomer kompliceras ytterligare av deras relativt stora storlekar och väsentliga inverterade och direkta upprepningar.
Traditionella protokoll för organellär isolering, DNA-rening och efterföljande genom E-sekvensering är ofta besvärlig och kräver stora volymer vävnadsinmatning, med flera gram uppåt i hundratals gram unga bladvävnader som är nödvändiga som utgångspunkt 11 , 12 , 13 , 14 , 15 , 16 , 17 . Detta gör organellärgenomsekvensering otillgänglig när vävnaden är begränsad. I vissa situationer är frömängderna begränsade, till exempel när det är nödvändigt att sekvensera generationsbaserade eller i manliga sterila linjer som måste bibehållas genom korsning. I dessa situationer kan organellär DNA renas och sedan utsättas för helgenomförstärkning. Hela genom-amplifiering kan emellertid introducera signifikant sekvenseringsförspänning, vilket är ett speciellt problem vid bedömning av strukturell variation, sub-genomiska strukturer och heteroplasminsnivåer> 18. Nya framsteg i bibliotekets förberedelse för kortläsning av sekvenseringsteknologier har övervunnit låga ingångsbarriärer för att undvika helgenomförstärkning. Till exempel tillåter Illumina Nextera XT-biblioteksberedningssatsen så lite som 1 ng DNA att användas som ingång 19 . Standardbiblioteksberedningar för långlästa sekvenseringsapplikationer, såsom PacBio eller Oxford Nanopore-sekvenseringsteknologi, kräver emellertid fortfarande en relativt hög mängd ingångs-DNA, vilket kan utgöra en utmaning för organellär genom-sekvensering. Nyligen har nya användarbaserade, långläsna sekvenseringsprotokoll utvecklats för att reducera ingångsmängderna och för att underlätta genomsekvensering i prover där man erhåller mikrogrammängder av DNA svårt 20 , 21 . Att erhålla högmolekylära vikt, rena organellära fraktioner för att mata in i dessa bibliotekspreparationer är emellertid en utmaning.
Vi sökte tO jämföra och optimera organellära DNA-anriknings- och isoleringsmetoder som är lämpliga för NGS utan behov av hel-genom-amplifiering. Specifikt var vårt mål att bestämma bästa praxis för att berika för organellärt DNA med hög molekylvikt från begränsade utgångsmaterial, såsom ett prov av ett blad. Detta arbete presenterar en jämförande analys av metoder för att berika för organellär DNA: (1) ett modifierat, traditionellt differentialcentrifugeringsprotokoll kontra (2) ett DNA-fraktioneringsprotokoll baserat på användningen av en kommersiellt tillgänglig DNA-CpG-metylbindningsdomänprotein 22 applicerad på växtvävnad 23 . Vi rekommenderar bästa praxis för isolering av organellär DNA från vävnadslövvävnad, som lätt kan utvidgas till andra växter och vävnader.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. Generering av växtmaterial för organellärisolering och DNA-extraktion
- Standardväxt av veteplanter
- Växtfrön i vermikulit i små, fyrkantiga krukor med 4-6 frön per hörn. Överföring till växthus eller växthus med 16 h ljuscykel, 23 ºC dag / 18 ºC natt.
- Vattna växterna varje dag. Gödda plantorna med ¼ tsk granulärt 20-20-20 NPK-gödselmedel vid spiring och vid 7 dagar efter spiring.
- Alternativ etiolering av veteplanter
- Följ steg 1.1, men lägg krukorna i en mörk tillväxtkammare, 23 ° C under 16 h / 18 ºC under 8 h. Alternativt täcka växterna i växthuset ( t.ex. med en förvaringsbehållare, men rätt ventilation måste bibehållas).
- Tillväxt och vävnadssamling
- Växa växterna i 12-14 dagar. För mest vetegenotypS, 75-100 plantor ger omkring 10-12 g vävnad, vilket är tillräckligt för två organella extraktioner med hjälp av differentialcentrifugeringsmetoden (avsnitt 2); Endast en växt är nödvändig om man använder DNA-CpG-metyleringsbaserad pulldown-tillvägagångssätt för fraktionering av organellär från kärn-DNA (avsnitt 3).
- Om man använder differenscentrifugeringsmetoden samlar man vävnad fräsch och fortsätter omedelbart för att bearbeta proven, som beskrivs i avsnitt 2.
- Om man använder CpG-metyl-pulldown-tillvägagångssättet skör 20 mg sektioner av ung bladvävnad i mikrocentrifugrör (använd antingen standardvuxen eller etiolerad vävnad, se representativa resultat ). Snapfrysning på flytande kväve och frys vid -80 ºC tills användning. Fortsätt till pulldownfraktionen av DNA, såsom beskrivs i avsnitt 3.
2. Metod nr 1: DNA-extraktion med differentialcentrifugering (DC)
OBS: diffErentiellt centrifugeringsprotokoll modifierades från två publikationer som optimerade betingelser för att isolera båda organellerna men berika för mitokondrier 17 , 24 . Det resulterande protokollet är mindre tidsintensivt och använder färre giftiga kemikalier än tidigare metoder. Specifikt gjorde vi modifieringar av buffertarna och tvättsteg, inklusive tillsats av polyvinylpyrrolidon (PVP) till STE-extraktionsbufferten och eliminering av slutstvättsteget i NETF-buffert, som innehåller natriumfluorid (NaF).
Varning: Beredningen och användningen av STE-bufferten ska utföras under en kemisk avloppsugn med lämplig personlig skyddsutrustning, eftersom denna buffert innehåller 2-merkaptoetanol (BME).
- Saker att göra innan du börjar
- Se till att all utrustning är extremt ren och autoklavera eventuell utrustning som kan autoklaveras ( t.ex. slipcylindrar, höghastighetscentriFuge rör, etc. ).
OBS! Filtertips rekommenderas för alla steg som kräver pipettering för att undvika korskontaminering. - Se listan över nödvändig utrustning och reagens och förbered de nödvändiga buffertarna och arbetslagren för metod nr 1 ( tabell 1 ). Kyl de kryogena slipblocken till -20 ºC och rotorerna och buffertarna till 4 ºC, sätt mikrocentrifugen till 4 ºC och sätt på ett 37 ºC vattenbad.
- Se till att all utrustning är extremt ren och autoklavera eventuell utrustning som kan autoklaveras ( t.ex. slipcylindrar, höghastighetscentriFuge rör, etc. ).
- Isolering av organeller
- Skala 5 g färskvävnad och skölj den i kallt, sterilt vatten i en kyld bägare på is.
OBS! Håll alltid proverna på is under alla operationer och transporter till och från centrifugerna, gaskåporna etc. Alternativt kan du arbeta i ett kallrum om det finns tillräckligt med utrymme och utrustning för att utföra protokollet. - Använd sax, skär bladvävnad i ~ 1 cm-stycken direkt i ett 50 ml rör som innehåller två keramiska slipningarcylindrar.
OBS: Rengör eller byt sax mellan prov för att undvika korskontaminering. - Om det inte finns någon vävnadshomogenisator, använd en mortel och pestel och följ för att ersätta steg 2.2.4 - 2.2.9.
- Skär bladvävnaden i en förkyld murbruk på is. Mala proverna i 2 - 3 min i 15 ml STE (i avloppsugan).
- Häll av bufferten (lämna vävnaden i morteln) genom en tratt som innehåller ett lager av för-våt, steril filtreringsduk (~ 22 till 25 μm porstorlek, se huvudprotokollet för detaljer) till ett annat 50 ml rör . Tillsätt ytterligare 10 ml STE till mortel och pistel och homogenisera igen.
- Häll den homogeniserade vävnaden och bufferten i samma tratt. Skölj mortel och pestle med 10 ml STE och häll det i tratten. Kram och vrid ut filtreringsduken i traven för att återställa så mycket vätska som möjligt.
OBS! Byt handskar mellan prov för att undvika korskontaminering. Fortsätt med proffsTocol i steg 2.2.10.
- Tillsätt 20 ml STE (i spishällen) till varje 50 ml rör.
- Placera proverna i förkylda kryogena slipblock i en vävnadsslipningsapparat och slipa proven i 2 x 30 s vid 1 750 rpm. Rotera provpositionerna och placera proven på is i ~ 1 min mellan grindarna.
OBS: En mortel och pestle, mixer eller annan vävnadsslipning / homogeniseringsanordning kan användas i detta steg. Varje metod kommer emellertid att påverka den resulterande DNA-kvaliteten i olika grader, och därför bör DNA-längd och kvalitet bedömas innan de fortsätter med applikationer nedströms. - Sätt i en tratt i ett rent 50 ml rör placerat i is. Placera ett lager av filtreringstyg i tratten och förtorka den med 5 ml STE. Kassera inte genomströmningen.
- Häll den homogeniserade vävnaden i tratten. Skölj slipröret med 15 ml STE, repetera och vänd röret för att skölja väggarna och locket och häll i funnel.
- Ta försiktigt bort de keramiska stenarna och kläm sedan och vrid ut filtreringsklattan i tratten.
OBS! Byt handskar mellan prov för att undvika korskontaminering. - Vik rörkapselen med parafilm för att undvika spill. Centrifugera vid 2000 xg i 10 min vid 4 ºC.
- Försiktigt aspirera supernatanten med hjälp av en serologisk pipett (undvik att störa pelleten) och placera den i ett centrifugrör med 50 ml höghastighetståg (om rören inte har täta tätningspackningar, linda rörkapselen med parafilm för att undvika spill). Kassera pelletsna.
- Balansera rören till inom 0,1 g med STE och centrifugera den erhållna supernatanten i 20 min vid 18 000 xg och 4 ºC. För att balansera rören, placera en liten bägare is på balans, tara skalan och väga prov på is för att hålla dem kalla. Alternativt, använd en balans och en avloppsugn i ett kallrum.
- Kassera supernatanten. Tillsätt 1 ml ST till pelleten och försiktigt suspendera ossI en mjuk pensel. Tillsätt 24 ml ST (slutvolym 25 ml) och blanda / virvel ( dvs tryck på penseln på rörets sida för att avlägsna all vätska).
- Balansera rören till inom 0,1 g med ST. Centrifugera i 20 min vid 18 000 xg och 4 ºC. Under tiden förbereder DNaseI-lösningen (se tabell 1 för lager- och arbetslösningsuppskrifter). För varje prov gör du en 200 μl alikvot i ett 1,5 ml rör.
- Kassera supernatanten, fläck röret och återupplösa pelleten (fortfarande i ett centrifugrör med hög hastighet) i 300 μL ST med en mjuk pensel. Placera penseln i det tidigare beredda 1,5 ml röret innehållande 200 μl DNaseI-lösning och vrida penseln för att avlägsna eventuell kvarvarande pellets fast i borsten. Pipettera DNaseI-lösningen tillbaka i höghastighetscentrifugröret och svälj försiktigt för att blanda.
- Inkubera vid 37 ° C i 30 minuter i ett vattenbad (linda parafilmen runt toppen av röret för att förhindra kondensationsläckageG in i locket). Blanda försiktigt genom att virvla 2 gånger under inkubation.
- Försiktigt pipetterar pelletsblandningen ur röret med hjälp av en pipettspets med en bred öppning och placerar den i ett 1,5 ml bindemedel. Tillsätt 500 μL 400 mM EDTA, pH 8,0, till centrifugröret med hög hastighet och försiktigt pipett för att få allt restpelleten ut ur röret. Överför EDTA till samma 1,5 ml lågbindningsrör som pelletsblandningen och blanda försiktigt genom inversion.
- Centrifugera vid 18 000 xg i 20 min vid 4 ºC. Kassera supernatanten, blott röret och använd genast för DNA-isolering. Frysta pellets vid -20 ºC vid behov, men detta kan resultera i en avkastningsminskning, eftersom resterande DNaseI kan försämra provd DNA om det inte omedelbart behandlas.
- Skala 5 g färskvävnad och skölj den i kallt, sterilt vatten i en kyld bägare på is.
- DNA-extraktion från isolerade organeller med användning av ett kommersiellt kolonnbaserat tillvägagångssätt
OBS! Se kithandboken för hela protokollet 25 , och se nedan för ändringar. PrÖkning direkt från organellärisolering till DNA-extraktion är att föredra. Upprepad frysning och upptining kommer att minska DNA-fragmentstorlekarna och leda till DNA-nedbrytning med kvarvarande DNaseI. Begränsa vortexing eller kraftig pipettering, eftersom detta kan skjuva DNA. Användningen av mikrocentrifugrör med låg bindning rekommenderas för att maximera DNA-återhämtningen.- DNA-extraktionsprocedur
OBS! Läs det detaljerade kommersiella protokollet 25 innan du börjar att se till att buffertarna är korrekt lagrade / lagrade och att spolningskolonnens procedurer förstås.- Tillsätt 180 μL buffert ATL direkt i röret med pelleten (upptinat om det tidigare är fryst och jämviktat till rumstemperatur på bänkskivan).
- Fortsätt med steg 3 i protokollet för "DNA-reningar från vävnader" i kithandboken med följande modifieringar: en 30-min-lysis i steg 3 inkluderar den valfria RNase A-digestionen och elueras i 3 x 200 | il AE ( Var och en i en seParate tube och sedan kombinera elutions).
- Spara en alikvot (minst 20 μl) för qPCR (se steg 4.1). För att kvantifiera innan du koncentrerar dig, spara ytterligare 1 μL för kvantifiering med hög känslighet.
- Fortsätt med provkoncentration om så önskas.
- DNA-extraktionsprocedur
- Provkoncentration med kommersiella filterenheter
OBS! Se handelsprotokollet 26 för mer information. Beroende på nedströmsanvändningen kan det inte vara nödvändigt att utföra provkoncentration ( t.ex. för slutpunkts-PCR- och qPCR-applikationer). För NGS-bibliotekskonstruktion är det emellertid sannolikt nödvändigt att koncentrera utspädd organellärt DNA erhållet efter DNA-extraktion.- Koncentrations kolonnförfarande
- Förväg väga (se tabell 2 ) den tomma filterenheten (utan ett rör) på ett rent stycke vägningspapper på en digital analysbalans. Spela in vikten.
- PiPette de kombinerade elutionerna in i filterenheten och väga noga igen.
OBS! Den kommersiella manualen 26 säger att maximal volym för filterenheten är 500 μL, men upp till 575 μL kan läggas till enheten samtidigt utan överflöde. - Placera den fyllda filterenheten försiktigt i ett rör (medföljer kolumnerna). Centrifugera vid 500 xg för önskad tid för att uppnå den erforderliga koncentratvolymen. För en provvolym på ~ 575 μL resulterar en 20-minuters spin normalt i en koncentratvolym av 15-30 μL.
- Ta bort filterenheten från röret och väga igen. Använd tabellen för att bestämma om önskad koncentratvolym har uppnåtts. Om inte, centrifugera igen vid 500 xg under en kortare tid och väga igen; Upprepa tills önskad koncentratvolym uppnås.
- Placera ett nytt rör (förses med kolumnerna) över filtrets enhet och invertera. Centrifugera i 3 minuter vid 1000 xg för att överföra coNcentrera till röret.
- Bestäm volymen återhämtad. Detta kommer normalt att vara ~ 3 - 5 μL mindre än den beräknade volymen, på grund av filterretention. Om det är överkoncentrerat, späd med sterilt vatten eller TE för att uppnå den önskade volymen.
- Kvantifiera DNA genom att använda högkänslig kvantifiering (enligt tillverkarens instruktioner).
- Koncentrations kolonnförfarande
3. Metod nr 2: Metylfraktionering (MF) -metod för att berika för organellär DNA från totalt genomiskt DNA
OBS: Detta protokoll modifierades från ett användarutvecklat DNA-extraktionsprotokoll för genomiskt tips-kit för växter och svampar 27 och det kommersiella Microbiome DNA Enrichment Kit-protokollet 28 . I teorin kan vilket DNA-isolationsprotokoll som ger DNA med hög molekylvikt användas för neddragningen. För kortläsning av sekvensering är varje extraktion som ger övervägande> 15 kb fragment tillräcklig för användning vid utlösningen. För loNg-läst sekvensering kan större fragment vara önskvärda. Därför optimerade vi detta protokoll för att ge DNA med hög molekylvikt.
- Isolering av totalt DNA
OBS: Se listan över nödvändig utrustning och reagens och förbered de nödvändiga buffertarna och arbetslagren för metod nr 2 ( tabell 1 ). Tillsätt lyseringsenzymer till lysbuffertlagret för att göra lysisbuffertens arbetslösning. Slå på termomixern och sätt den till 37 ° C. Slå på vattenbadet till 50 ° C och sätt QF-buffert i badet. Placera 70% EtOH i frysen och sätt mikrocentrifugen till 4 ° C.- Total DNA-extraktion med kommersiella DNA-extraktionskolonner
OBS! Innan du börjar, läs den kommersiella handboken 29 för detaljerad information om användningen av anionbytarkolonnerna med gravitationen. Kolonnerna kan ställas in med hjälp av en specialstativ eller placeras över rören med hjälp av de medföljande plastringarna. Alla steg, inklusive gEnomic tips, bör tillåtas fortsätta med gravitation flöde, och resterande vätska bör INTE tvingas igenom.- Mala 20 mg frusen vävnad i flytande kväve i ett 2-ml lågbindningsrör med handhållna slippistlar avsedda för 2 ml rör.
- Tillsätt 2 ml lysisbuffertarbetslösning (rören blir mycket fulla).
- Inkubera i en termomixer vid 37 ° C under 1 timme med försiktig omröring vid 300 rpm. Om en termomixer inte är tillgänglig, är det lämpligt alternativ att inkubera på ett värmeblock och blanda genom försiktig blinkning var 15: e minut.
- Tillsätt 4 μl RNase A (100 mg / ml, slutkoncentration av 200 μg / ml). Invertera för att blanda och inkubera i en termomixer under 30 minuter vid 37 ° C med försiktig omröring vid 300 rpm.
- Tillsätt 80 μl proteinas K (20 mg / ml, slutkoncentration av 0,8 mg / ml), invertera för att blanda och inkubera i en termomixer i 2 h vid 50 ° C med försiktig omröring vid 300 rpm.
- Centrifugera i 20 minuter vid 4 ° C och 15.000 xg för att pelletera olösliga skräp.
- Medan proven är centrifugering, jämvikter kolonnerna med 1 ml buffert QBT och låt kolonnen tömma genom gravitationens flöde.
- Använd en pipettpip med brett borr för att omedelbart applicera provet (undvik pelleten) till den jämviktspaltiga kolonnen och låt den strömma helt genom kolonnen. Om provet blir grumligt, filtrera eller centrifugera igen innan det appliceras i kolonnen (se den kommersiella handboken för detaljer 29 ).
- När provet helt har gått in i hartset, tvätta kolonnen med 4 x 1 ml buffert QC.
- Suspension kolonnen över ett rent, 2 ml, bundet mikrocentrifugrör. Eluta genomiskt DNA med 0,8 ml buffert QF förvärmd vid 50 ° C.
- Precipitera DNA genom att tillsätta 0,56 ml (0,7 volymer elueringsbuffert) av rumstemperaturisopropanol till det eluerade DNA.
- Blanda genom inversion (10X) och centrifugera omedelbart i 20 minuter vid 15.000 xg och 4 ° C. VårdTa helt bort supernatanten utan att störa den glasartade, löst fastsatta pelleten.
- Tvätta den centrifugerade DNA-pelleten med 1 ml kall 70% etanol. Centrifugera i 10 minuter vid 15 000 xg och 4 ° C.
- Ta försiktigt bort supernatanten (var försiktig med detta steg också) utan att störa pelleten. Lufttorka i 5-10 min och resuspendera DNA: t i 0,1 ml elueringsbuffert (EB). Lös DNAet över natten vid rumstemperatur. Undvik pipettering, vilket kan skjuva DNA: n.
- Kvantifiera proverna med hjälp av en högkänslig DNA-kvantifieringsanalys (enligt tillverkarens instruktioner).
- Total DNA-extraktion med kommersiella DNA-extraktionskolonner
- Beadbaserad fraktionering av metylerat och omättat DNA
ANMÄRKNING: En nyligen publicerad publikation visade användningen av ett kommersiellt tillgängligt kit 28 som utnyttjar ett pulldown-tillvägagångssätt med användning av ett CpG-specifikt metylbindande domänprotein fuserat till humant IgG Fc-fragment (MBD2-Fc-protein) till fraktionÅt växtorganellärgener (omättat) från kärngenom (högt metylerat) halt 23 . Fraktioneringseffektivitet i vetexemplar testades inte tidigare med användning av detta kommersiella MF-kit 28 .- Saker att göra innan du börjar
- Färskberedning 80% etanol (minst 800 μl per reaktion). Ställ 5x bind / tvättbuffert för att tina på is och förbered 5 ml 1x buffert per prov (utspädd 5X-buffert med sterilt, nukleasfritt vatten och fortsätt isen under protokollet).
- Förbereda MBD2-Fc proteinbundna magnetiska pärlor
- Förbered det önskade antalet pärlsatser. Skala reaktionerna för att använda mellan 1 och 2 μg totalt insignal DNA, vilket kräver 160-320 μl pärlor. Observera att reaktionerna som anges nedan är för 1 μg av totalt inmatnings-DNA, så de behöver 160 μl pärlor. Skala reaktionerna efter behov.
- Använda brettborrspetsar, pipett försiktigt protein A Magnetic BEad slammar upp och ner för att skapa en homogen suspension. Som ett alternativ rotera röret med pärlor försiktigt i 15 minuter vid 4 ° C.
OBS: Vortexa inte pärlorna. - Fortsätt med anvisningarna enligt tillverkarens anvisningar 28 .
- Fånga metylerat nukleärt DNA
- För varje enskilt prov tillsätt 1 μg ingångsd DNA till ett rör innehållande 160 | j, l MBD2-Fc-bundna magnetiska pärlor.
- Lägg till 5x bind / tvättbuffert, beroende på vad som är lämpligt, med tanke på volymen av DNA-ingångsprovet för en slutlig koncentration av 1x (volym 5x bind / tvättbuffert för att lägga till (μL) = volym ingångsd DNA (μL) / 4). Pipettera provet upp och ner några gånger för att blandas med en pipettpip med brett borrning.
- Vrid rören vid rumstemperatur i 15 minuter. Försiktigt pipettera proverna med en brettborrspetspets och flip proverna 2-3 gånger genom inkubationen för att förhindra kollapsning av pärlor.
OBS: pipetteringen och flickanNg är kritisk för att säkerställa effektiv pulldown av det metylerade DNA.
- Samla berikad, icke-metylerad organellär DNA
- Snurra snabbt röret som innehåller DNA-och MBD2-Fc-bunden magnetisk pärlblandning. Placera röret på en magnetstativ i minst 5 minuter för att samla pärlorna till rörets sida. Lösningen ska vara tydlig.
- Använda brettborrspetsar, försiktigt avlägsna den rensade supernatanten utan att störa pärlorna. Överför supernatanten (innehåller icke-metylerat, organellärberikat DNA) till ett rent, lågbindat, 2 ml mikrocentrifugrör. Förvara detta prov vid -20 eller -80 ° C, eller fortsätt direkt till steg 3.2.6 för rening.
- Eluta fångat nukleärt DNA från de MBD2-Fc-bundna magnetiska pärlorna
- Om kärnfraktionen också är önskvärd följer tillverkarens instruktioner 28 för att eluera det kärn-DNA från de MBD2-Fc-bundna magnetiska pärlorna; Rena som beskrivet i steg 3.2.7.
- Beadbaserad nukleinsyrarening
- Se till att reningspärlorna är vid rumstemperatur och blandas noggrant. Fortsätt med protokollet enligt anvisningarna i manualen för MF-kit 28 .
OBS: Provet kan nu användas för NGS-bibliotekskonstruktion eller en annan nedströmsanalys.
- Se till att reningspärlorna är vid rumstemperatur och blandas noggrant. Fortsätt med protokollet enligt anvisningarna i manualen för MF-kit 28 .
4. Provkvantificering och kvalitetskontroll
- QPCR-analys för att utvärdera organellär anrikning
OBS: De qPCR-reaktions- och analysparametrar som listas här är konstruerade för användning på en Roche LightCycler 480 och kan behöva justeras för olika utrustningar och reagenser. Om qPCR är otillgänglig kan slutpunkts-PCR och visualisering på en agarosgel användas som en kvalitativ mätning av provrenhet med användning av samma primers och betingelser som beskrivs här. Amplicon storlekar kommer att vara ~ 150 bp för alla primersatser. Se tabell 3 för primer-sekvensenCes och parningar.- QPCR reaktion setup
- För att ställa in en individuell 20 μl qPCR-reaktion pipetteras följande försiktigt i en brunn i en 96-brunns qPCR-platta: 10 μL 2x SYBR Green I Master; 2 μl av 10 μM framåt och bakre primerblandningen (för en slutlig koncentration av 0,5 | im); 2 μl mall (inom intervallet av standardkurvan); och 6 | il av sterilt, nukleasfritt H 2 O. För att minska pipetteringsfel, är det föredraget att göra en huvudblandning med alla reaktionskomponenter förutom mall. Lägg till mastermixen på qPCR-plattan och lägg sedan till mallen av intresse för varje brunn. Tre tekniska replikat för varje prov bör utföras för att minimera effekterna av pipetteringsfel.
OBS: I slutändan jämförs kvoten mellan kärnor och organellära kvantifieringscykler mellan proverna, så små skillnader i koncentration är acceptabla. DNA-koncentrationerna bör dock vara ungefär inom intervallet eaCh andra. - Tät plåten med en högkvalitativ qPCR-tätningsfilm. Vortexa proverna försiktigt och var försiktig så att inga bubblor skapas. Snurra plattan kort i 2 minuter vid 4 ° C för att samla provet och eliminera eventuella små bubblor.
- Placera plattan i maskinen. Kör qPCR-programmet enligt de riktlinjer som anges nedan.
- För att ställa in en individuell 20 μl qPCR-reaktion pipetteras följande försiktigt i en brunn i en 96-brunns qPCR-platta: 10 μL 2x SYBR Green I Master; 2 μl av 10 μM framåt och bakre primerblandningen (för en slutlig koncentration av 0,5 | im); 2 μl mall (inom intervallet av standardkurvan); och 6 | il av sterilt, nukleasfritt H 2 O. För att minska pipetteringsfel, är det föredraget att göra en huvudblandning med alla reaktionskomponenter förutom mall. Lägg till mastermixen på qPCR-plattan och lägg sedan till mallen av intresse för varje brunn. Tre tekniska replikat för varje prov bör utföras för att minimera effekterna av pipetteringsfel.
- QPCR-reaktionsparametrar
OBS! Dessa är standardparametrar, förutom förhöjningscykeln i förstärkningssteget. Justera denna inställning för att rymma specifika primers om de som används skiljer sig från de primers som presenteras i detta protokoll.- Förinkubera vid 95 ° C i 5 min, med en ramphastighet av 4,4 ° C / s.
- Utför 45 amplifieringscykler av (1) 95 ° C under 10 s, med en ramphastighet av 4,4 ° C / s; (2) 60 ° C i 20 s, med en ramphastighet av 2,2 ° C / s; Och (3) 72 ° C i 10 s, med en ramphastighet av 4,4 ° C / s (data förvärvad under (3)).
- Använd en optioNal smältkurva cykel av 95 ° C i 5 s med en ramphastighet av 4,4 ° C / s; 65 ° C i 1 min, med en ramphastighet av 2,2 ° C / s; Och 97 ° C, med ett kontinuerligt förvärvsläge.
- Använd en kylcykel på 40 ° C i 30 s, med en ramphastighet på 1,5 ° C / s.
- Analysparametrar
- Välj SYBR-mallen. Kontrollera programparametrarna i Experiment-knappen. När plattan är laddad kan analysen startas, och inställningarna kan justeras medan analysen körs.
- Tilldela prov med hjälp av provredigeraren. Välj Abs Quant som arbetsflöde och ange proven som okänd, standard eller negativ kontroll. Designate replikerar och fyller i provnamnen på den första av varje replikat. Lägg till koncentrationer och enheter till standarderna.
- Ställ in subsets för analys; Dessa tilldelas i deluppsättningsredigeraren.
- För analys, välj Abs Quant / 2nd Derivative Max från listan "skapa ny analys".Importera externt sparade standardkurvan (om tillämpligt) och klicka sedan på beräkna; Rapporten innehåller de valda uppgifterna.
- För att utföra exakt absolut kvantifiering för bestämning av kopiantal eller koncentration, använd en standardkurva som är representativ för provet som testas (t ex organellär DNA isolerat från ovanstående metoder). Eftersom mängden mitokondriellt DNA som krävs för att förbereda en standardkurva är för hög för att uppnås med en rimlig mängd vävnad, utnyttjar inte kopiantalberäkningar som tillhandahålls av mjukvaran, utan undersöker i stället gränsvärdesvärden (Cp) för bestämning av den relativa anrikningen Av organellar jämfört med nukleärt DNA i proverna. Jämför dessa relativa mängder med de totala genomiska DNA-värdena (se representativa resultat ). Testprimeringseffektivitet på fem 1:10 utspädningar av totalt genomiskt DNA från helt ljusvuxna, två veckor gamla veteplanter (representativa effektiviteter rapporterade iE legenden i figur 2 ).
- QPCR reaktion setup
- Pulsfältgelelektrofores (PFGE)
OBS! Detta protokoll är baserat på tillverkarens riktlinjer för att utföra PFGE för att lösa DNA med hög molekylvikt. Se materialtabellen.- Förbereda gelén och proven
- Följ riktlinjerna för gel- och provberedning och anpassa dem till det tillgängliga systemet.
- Kör parametrarna
- Följ anvisningarna för uppställning av elektroforesystemet och använd följande parametrar: starttid för omkopplingstid på 2 s, slutlig omkopplingstid på 13 s, körtid på 15 h och 16 min, V / cm 6 och ingående vinkel på 120 ° .
- Färga och bilda gelén
- Färga gelén med ett valfritt färgämne ( t.ex. etidiumbromid eller ett lämpligt alternativ) och bild med ett lämpligt geldokumentationssystem.
- Förbereda gelén och proven
- Använd 1 ng DNA som input för DNA Library Prep Kit, enligt tillverkarens instruktioner.
- Streckkod och samla proverna för sekvensering i en enda running. Utför sekvensering enligt tillverkarens riktlinjer.
OBS! Parametrar och sekvenseringsparametrar kan ändras beroende på arten av intresse, önskad täckningsnivå och plattformen som används för att sekvensera biblioteken. Exempelvis har en HiSeq-lane väsentligt mer effekt än en MiSeq-lane, så många fler prover kan multiplexeras. Sekvensera en mindre delmängd av prover för att bestämma om täckningsnivåerna hos organellärgenerna är tillräckliga för nedströmsanalys.- Undersök läskvaliteten med FastQC 31 för att bestämma graden av trimning och filtrering som krävs för data.
- Trim och filtrera den råa läsningen med Trimmomatic 32 eller ett annat jämförbart program. Använd följande inställningar: ILLUMINACLIP 2:30:10 (för att ta bort adaptrar), LEADING 3, TRAILING 3, SLIDINGWINDOW 4:10 och MINLEN 100.
- Map kvaliteten-filtrerades och adapter-trimmade parade-änden (PE) läser för kinesiska Spring mitokondrie (NCBI Reference Sequence NC_007579.1 33), kloroplast (NCBI Reference Sequence NC_002762.1 34), och nukleära 35 referens genomen med användning Bowtie2 36, Med följande inställningar: -I 0 -X 800 - känslig.
- Konvertera sam alignment-filer till bam-format (samtools) och sortera bam-filerna. Använd bam-filerna för att beräkna genomsäckande täckning och täckning per bädd med bedtools. Visualisera resultaten med R-plot-funktionen.
- Saker att göra innan du börjar
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
De protokoll som presenteras i detta manuskript beskriver två separata metoder för att berika för organellär DNA från växtvävnad. Villkoren som presenteras här återspeglar optimering för vetevävnad. En jämförelse av nyckelsteg i protokollen, obligatorisk vävnadsingång och DNA-utmatning beskrivs i figur 1 . Stegen i det DC-protokoll som vi testat följer liknande förhållanden som de som beskrivits tidigare ( Figur 1A ). Skördad vävnad måste behandlas färskt och utsättas för differentiell centrifugering och / eller gradient för att isolera intakta organeller. Nukleär DNA elimineras innan organellerna lyseras och slutligen extraheras DNA och används för nedströmsapplikationer. I motsats till detta, i MF-protokollet kan växtvävnad skördas och lagras före användning, och intakta organeller är inte nödvändiga. Istället fraktioneras kärn- och organellär DNA från totalt gDNA baserat på DNA-metyleringsstatusen. Båda protokollen ger ungefär lika stora mängder organellär DNA ( figur 1C ). När det gäller total organellär DNA-produktion relativt vävnadsinmatning är MF-protokollet fördelaktigt när vävnaden är begränsad, eftersom ett litet prov från en enda växt kan användas och växten får tillåtas växa för vidare analys. Vanligtvis krävs i DC-protokoll alla luftvävnader från många plantor, och dessa växter kasseras. DC-metoden kan emellertid optimeras för att specifikt berika för en organell typ över den andra, vilket inte är möjligt med MF-tillvägagångssättet. Det är värt att nämna att den totala tiden för varje protokoll är ungefär likvärdig, även om det finns mindre hands-on-tid i MF-tillvägagångssättet.
Båda metoderna berikar för organellär DNA, om än med olika proportioner av mitokondrier och plastidsekvenser:
"> Mycket låga mängder renat organellärt DNA erhålls från båda metoderna (i storleksordningen 50-100 ng, Figur 1C ). För att bedöma nivåerna av organellärt genomanrikning och kärngenomförorening i DNA isolerat från både DC och MF Metoder, en qPCR-analys användes. I denna analys användes de relativa överflödena av tre amplikoner ( dvs nukleinspecifika , ACTIN , mitokondrialspecifika, NAD3 ; Kloroplastspecifik, PSBB ) bedömdes i totalt genomiskt DNA och den organella DNA-fraktionen erhölls från båda metoderna ( Figur 2 ). Kvantifieringscykelvärden (C q ) undersöktes för varje prov ( Figur 2A ) och eftersom C q definieras som PCR-cykeln, vid vilken fluorescensen från målförstärkningen ökar över bakgrundsfluorescensnivån, har C q och målöverflöd en omvänt förhållande. IDC provet, Cq av NAD3 och PSBB är respektive ~ 17 och ~ 15 cykler tidigare än aktin (som har en C-q på ~ 36) (se figur 2B för C q-värden och anrikningsnivåer). Detta motsvarar teoretiska 167.181- och 47.790-faldiga berikningar för respektive NAD3 respektive PSBB , I förhållande till ACTIN i DC-provet ( Figur 2B , se legenden i Figur 2 för beräkningen). I det totala genomiska DNA-provet är vikningsberikningarna för NAD3 och PSBB i förhållande till ACTIN endast 158 respektive 10 701. Det är inte överraskande att hitta en högre överflöd av de organellära amplikonerna i förhållande till det kärnamplikon i totalt genom-DNA, med tanke på att organellärgenerna existerar i större exemplarantal per cell än kärngenomet 37 och att antalet organeller peR cellen kan skilja sig beroende på vävnadstyp eller utvecklingsstadiet 38 , 39 . Sammantaget indikerar data att DC-metoden företrädesvis berikar för mitokondrier, vilket kan förväntas, eftersom centrifugeringshastigheter optimeras för att selektivt isolera mitokondrier och reducera kärn- och kloroplast "kontaminering".Den omättade fraktionen av MF totalt gDNA visar också väsentlig berikning av båda organellära ampliconerna och förväntas bibehålla de nativa relativa mängderna av dessa mål. Foldanrikningarna för NAD3 och PSBB i förhållande till ACTIN i den ommetylerade fraktionen är 20.551 respektive 1.703.253 ( Figur 2A och 2B ). I den metylerade fraktionen är vikförgreningarna för NAD3 och PSBB relativt ACTIN 31 respektive 823 indiCating att MBD2-Fc-protein är mycket effektivt vid neddragning av metylerat nukleärt DNA. Eftersom kloroplast amplicon har en högre mängd än mitokondrie amplicon i totalt genomiskt DNA (~ 6 C q tidigare), metylerad fraktion (~ 5 C q tidigare) och icke-metylerad fraktion (~ 6 C q tidigare), tyder detta på att Ursprunglig överflöd av dessa amplikoner förändras inte väsentligt genom MDB2-rullning. Vi fokuserar här på den icke-metylerade (organellära) fraktionen på grund av intresset att sekvensera dessa genomer specifikt. Om emellertid nukleärt genom är det primära intresset, skulle MF och efterföljande sekvensering av den metylerade fraktionen ge en mycket högre kärnometomtäckning än total genomisk DNA-sekvensering på grund av minskningen av organellär DNA-"kontaminering".
Det är värt att notera att om qPCR inte är tillgänglig, ger slutpunkts-PCR (med samma primers som för qPCR) kvaliténTiv bedömning av organellär renhet. I detta fall kommer rena organellära DNA-prover att visa amplifiering för mitokondriella och plastid-ampliconerna, men ingen detekterbar amplifiering av nukleär amplicon på agarosgelén, medan totalt genomiskt DNA visar amplifiering för alla tre primeruppsättningar, vilket demonstrerats i tidigare studier 11 , 12 .
Organellär DNA isolerat från båda metoderna är lämpligt för NGS:
Trimmad och rengjord PE sekvensering läser (se steg 4.3) mappades till tidigare publicerade veteorganella referensgenom, och mängden läsningar som användes för kartläggning av varje prov varierade från ~ 800 000 till 1 100 000 läser ( Figur 3I ). Resultat från kartläggning de novo Illumina-sekvensering läser till tillgängliga vetekloroplast och mitokondria genomer överensstämmer med qPCR resUlts, med DC-metoden som ger DNA som är mer berikat i mitokondriellt DNA ( Figur 3A och 3B , ~ 80% och ~ 10% av läser kartan till respektive mitokondriella (mt) och kloroplast (cp) genomerna) och MF-metoden Vilket ger DNA som sannolikt återspeglar den ursprungliga överflöden hos de två organella-genomerna ( figurerna 3A och 3B , ~ 20% och ~ 80% av läser kartan till respektive mt- och cp-genomerna). I båda metoderna överstiger den teoretiska täckningen (se legenden i figur 3 för beräkningen) av båda veteorganella genomerna över 100X täckning (och varierar upp till ~ 2.000X täckning för kloroplastgenomet i den ommetylerade fraktionen från MF-metoden), jämn När 12 bibliotek multiplexeras ( Figur 3C och 3D ; de 6 bibliotek som ingår i denna analys slogs samman med ytterligare 6 bibliotek för en separat analys, för totalt 12 bibliotekSammanslagna i en enda sekvenseringsfält). En mer detaljerad bild av täckningen uppnåddes genom att undersöka fraktionen av genomet som omfattas av specifika djup samt vid täckningsnivåer per bas ( Figur 3E -3I ). För MF-metoden var den genomsnittliga per-bas-täckningen ~ 300 - 450X för mt-genomet och 4000-5000X för cp-genomet. För DC-metoden var den genomsnittliga per-basdäckningen ~ 900-10000 och ~ 500-700X för respektive mt- och cp-genomerna. Det fanns dock en liten del av både mt- och cp-genomerna som hade extremt låg eller hög täckning, och detta sågs i organellärt DNA härrörat från båda metoderna ( Figur 3I ). Regioner med högre än genomsnittlig täckning som sannolikt motsvarar regioner av homologi mellan organellärgenomerna och regioner med låg täckning kan indikera SNPs eller andra små varianter mellan de sorterade vi sekvenserade och de publicerade referenserna. Till stöd för denna uppfattning, dessa spikarAv hög täckning var mest uttalad för mt-DNA härledt från MF-metoden ( figurerna 3E och 3I ), troligen på grund av den höga täckningen av cp-genomet i denna metod. Otvetydigt är täckningen av cp-genomet mer ojämn i MF-metoden än DC-metoden ( Figur 3G och 3H ), vilket kan bero på små förspänningar i MBD2-Fc-pulldownen längs cp-DNA. Ytterligare experiment kommer att krävas för att avgöra varför detta är fallet. Oavsett att mt- och cp-genomerna hade relativt jämn täckning med båda metoderna och inga stora områden med saknad täckning, vilket kan demonstreras genom undersökning av fraktionen av genomer sekvenserade vid ett visst djup ( Figur 3E -3H ). Dessutom anses täckningsnivåerna för båda genomerna tillräckliga för nedströmsanalys, såsom variantanalys. Om det anses nödvändigt för analys av sällsynta varianter, minskning av numbeR av sammanslagna prover skulle uppnå större täckning. Alternativt kan ett mycket större antal prover samlas i en HiSeq-banan, samtidigt som det uppnår ännu större sekvenseringsdjup, om än vid ett offer i sekvenslängd, eftersom HiSeq-bibliotek är för närvarande begränsade vid PE150-längden i motsats till PE300 MiSeq-bibliotek.
För att undersöka nukleärgenoms kontamineringsnivåer med hjälp av en kartläggning, undersöktes PE-läsningskategorier. PE-läsningar kan kartläggas till ett referensgenom i olika konfigurationer. När man läser 1 och 2 riktar sig till referensen i ett huvud mot huvudet med ett visst "förväntat" avstånd mellan de två kompisarna (baserat på bibliotekets genomsnittliga insatsstorlek och typiskt angiven som en ingångsparameter i kartläggningsprogrammet ), Dessa PE läser sig att kartlägga "concordantly." I motsats till detta är "otillbörlig" kartläggning den situation där kompisar kartläggs med en mindre eller större än förväntad disÖverensstämmelse med referensgenomet eller kartan i alternativa konfigurationer (från topp till svans eller från svans till svans). Om endast en kompis riktar sig till referensgenomet, sägs det PE läsas att varken överensstämma eller obekvämt med referensgenomet. I alla tre avläsningskategorier kan PE-läsningar stämma överens med referensgenomet en eller flera gånger.
För både DC- och MF-isolerat organellärt DNA var läsning av kartläggning till mitokondriella genomet övervägande i den inriktade likvärdigt en tidskategorin ( Figur 4A ), medan läser kartläggs till kloroplastgenomet i relativt lika stora proportioner av överensstämmelse en gång och överensstämmande mer än En gång ( Figur 4B ), troligen på grund av de stora inverterade upprepningarna närvarande i kloroplastgenomet och även till de extremt höga täckningsnivåerna. Men färre PE läser kort till kärngenomet och kartläggs i stor utsträckning mer än en gång i aVarken överensstämmande eller oskäligt sätt ( dvs. endast en kompis kan kartlägga). Dessa är mest sannolika att kartlägga "off-target" till sekvenser i kärngenomet, vilka är homologa med organellärgenerna eller felaktiga regioner. Endast en mindre mängd avläser (<5%) som kartläggs till kärngenometet med överensstämmelse, vilket indikerar låga nivåer av kärngenomförorening i organellärt DNA isolerat från DC eller MF-metoden ( Figur 4C ), vilket också reflekteras av qPCR-resultaten ( Figur 2A ). Kärnfragmentet efter MBD2-Fc-pulldown från kinesiska vårens icke-etiolerade vävnader sekvenserades också för att bestämma hur effektiv pulldownen är vid avlägsnandet av omättat DNA. Mindre än 1% av läser i det kärnfragment-härledda biblioteket mappat till organellära referensgenom, medan ~ 45% av alla läser kartläggs till kärngenometet ( Figur 4 ). Dock läser de flesta kartor på ett oklanderligt sätt, wVilket sannolikt återspeglar de höga nivåerna av sammansättning och fragmentering i vetekärnreferensgenomet. Oavsett att resultaten tyder på att MBD2-Fc-pulldownen är mycket effektiv vid avlägsnandet av icke-metylerat organellärt DNA från metylerat nukleärt DNA. Det är värt att notera att eftersom den organellärberika DNA som härrör från dessa metoder innehåller en blandning av mitokondrier och kloroplastsekvenser och eftersom sekvenslikheter som härrör från forntida genöverföring mellan dessa organeller förblir i deras genomer, läser den korrekta uppgiften till den specifika Genomerna måste lösas bioinformatiskt.
Etiolationen av bladvävnad förändrar inte på ett rimligt sätt organiska överflöd:
Traditionellt föredras etiolerade vävnader för isolering av mitokondriell DNA för att minska nivåerna av fenoler och stärkelser, vilket kan störa extraktionN eller nedströms applikationer 13 . För att bestämma om organellärgenomrikningsnivåer kan förändras eller förbättras genom tillväxtbetingelser, utsattes både etiolerade och icke-etiolerade vävnader för MF-protokollet och sekvensering. Intressant förändrade etiolering inte märkbart andelen av läsningar som kartlades till organellärreferensgenomerna ( figurerna 3A och 3B ) eller per- basdäckningen ( figur 3I ) jämfört med icke-etiolerade betingelser. Vi isolerade också organellär DNA med differentiell centrifugering, med både etiolerade och icke-etiolerade vävnader, och liten skillnad i anrikning hittades mellan de olika vävnaderna med användning av qPCR (data ej visad). Detta tyder på att mer fysiologiskt relevanta icke-etiolerade vävnader kan användas för organellära sekvensstudier, utan någon märkbar förändring av anrikning.
Kvalitetskontroll föreslår detMF-DNA är mest lämpligt för långlästsekvensering:
Eftersom långlästsekvensering blir mer tillgänglig för forskare blir isoleringen av DNA med hög molekylvikt allt viktigare. För att utvärdera organellär DNA isolerat med antingen metod för intaktitet och kvalitet användes PFGE. Totalt genomiskt DNA migrerar typiskt som ett diffust smält i PFGE, och molekylvikten bestäms av protokollet och hur DNA lagras och hanteras efter extraktion. Det totala genomiska DNA som isolerats med genomiska tips bör överstiga 50 kb, vilket verifierades med användning av PFGE ( Figur 5 , fält 2). Det totala genomiska DNA från de genomiska tipsen används som inmatning i Microbiome Enrichment Kit för att fraktionera kärnan från organellär DNA. Den nukleära fraktionen som erhålls efter fraktionering minskar i storlek, men är fortfarande centrerad runt 50 kb ( Figur 5 , fält 4). Detta är inte suUnder förutsättning att den förhållandevis hårdare hanteringen av kärnfraktionen som eluering från MBD2-Fc-bundna pärlor kräver värme- och proteinas K-digestion. På grund av den begränsade massan kördes inte organellarfraktionen på PFGE, men efterföljande analys med TapeStation indikerade DNA> 50 kb (data ej visad). Det organellära DNA som erhålls med differentiell centrifugering har en medelmassa av ~ 20 kb, sannolikt orsakad av det förlängda organella isolationsprotokollet och den efterföljande kolonnbaserade DNA-extraktionen och koncentrationen. Gradientbaserad organellärisolering och alternativa DNA-extraktionsmetoder kan upprätthålla större DNA-fragmentstorlekar. Oavsett, DNA av den storlek som erhållits i detta protokoll kan användas för att generera 10- eller 15-kb-sekvensering läser om det tas hand om under bibliotekets beredning.
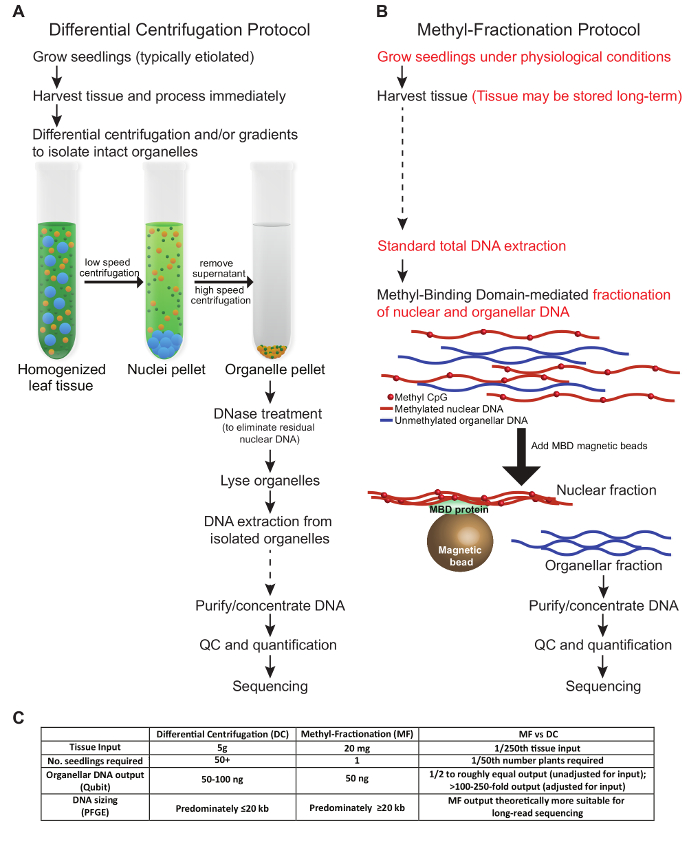
Figur 1: En jämförande vy av två metoDs att berika för plantorganellär DNA. Ett traditionellt DC-protokoll ( A ) står i motsats till MF-protokollet ( B ). Det rekommenderas att frysa och tina proverna. Steg där proven kan lagras på lång sikt anges dock med streckade pilar ( A och B ). Viktiga skillnader mellan protokollen markeras i rött ( B ). ( C ) Tabellen jämför metoderna i form av vävnadsinmatning, antal plantor som krävs, DNA-utmatning och resulterande DNA-storlek. Vänligen klicka här för att se en större version av denna figur.

Figur 2: Bedömning av kärn-DNA-kontaminering i organellär DNA-isolerad med användning av två metoder. (
( B ) Tabellen visar C q- värdena, som visas i diagrammet i ( A ) och vikningsanrikningen av organellära amplikoner i förhållande till ACTIN . * Vika anrikning = 2 (Cq ACTIN - Cq Target) . Formeln förutsätter en perfekt effektivitet av 2 för varje primeruppsättning, sedan den mindre deviatJon för varje primer som är inställd från 2 är försumbar och skulle få liten effekt på beräkningen och den övergripande trenden ( ACTIN = 1.961, NAD3 = 1.95 och PSBB = 1.989). Primereffektiviteten utvärderades genom att en standardkurva framställdes med en serie av fem 1:10 spädningar av totalt genomiskt DNA. Vänligen klicka här för att se en större version av denna figur.

Figur 3: Läs kartläggning och teoretisk täckning av kloroplast och mitokondriella genom. Procentandel av läser kortlagd till mitokondriella ( A ) eller kloroplast ( B ) kinesiska vårreferensgenomerna. Motsvarande teoretisk täckning av den kinesiska vår mitokondriella ( C ) eller kloroplast ( D ) referensgenoMes, med antagande av genomstorlekar på 450 respektive 135 kb, beräknade med hjälp av det totala läsnumret och procentandelen av läsning av kartläggning till de olika genomerna. Genomgående fördelning av täckning för organellär DNA från MF-metoden ( E och G ) eller DC-metoden ( F och H ). Uppgifterna i paneler E - H är från det kinesiska fjäderets etiolerade provet, men alla andra prover visade en liknande trend. ( I ) Genomsnittlig, lägsta och högsta per-bas täckning för alla prover i paneler A- D . Provetiketter inklusive "E" betecknar etiolerade prover, och "NE" betecknar icke-etiolerade prover. DC indikerar DNA isolerat med differentieringscentrifugeringsmetoden och omättat metylen indikerar DNA som är i den ommetylerade fraktionen efter pulldown med MBD2-Fc (MF-protokoll). Prover märkta "Chris" betecknar vete Triticum aestivum'Chris'. CS betecknar prover av vete Triticum aestivum 'Chinese Spring. Anm: På grund av sekvenshomologi mellan kloroplast, mitokondrier och kärnomgener som härrör från gammal genöverföring mellan organellärgenerna samt mellan organellära och nukleära genomer, kan en liten procentandel av råa läsningar kartlägga till multipla genomer. Dessutom, läser som inte kartlägger till antingen organellär referensgenomet är inte representerade i denna figur. Följaktligen uppgår inte procenttalen som visas här ( A och B ) 100%. Vänligen klicka här för att se en större version av denna figur.

Figur 4: PE-läsning av kartläggning till vete-kärngenomet. Andel av kategorier av PE Läs kartläggningstyper till mitokondriella (A) , kloroplast (B) eller kärnor (C) kinesiska vårreferensgenom. - E betecknar etiolerade prover och - NE betecknar icke-etiolerade prover. DC indikerar DNA isolerat med differenscentrifugeringsmetoden. Umetylerad indikerar DNA som ligger i den ommetylerade fraktionen efter pulldown med MBD2-Fc i MF-protokollet, och metylerad betecknar kärnfraktionen efter MBD2-Fc-pulldown. Prover märkta "Chris" betecknar vete Triticum aestivum 'Chris.' CS betecknar prover av vete Triticum aestivum 'Chinese Spring.' Omapplade läsningar visas inte. Vänligen klicka här för att se en större version av denna figur.
Oad / 55528 / 55528fig5.jpg "/>
Figur 5: Undersökning av DNA-kvalitet med användning av PFGE. Vete totalt genomiskt DNA (lane 2), veteorganellärt DNA erhållet från differentiell centrifugering (lane 3) och kärnfraktionen efter MF med MBD2-Fc-lindningsmetoden (lane 4) utsattes för PFGE på en 1% agarosgel med en 1 kb förlängd stege som används som markör (banor 1 och 5). Vänligen klicka här för att se en större version av denna figur.
| Buffertnamn | Recept | anteckningar | Metod |
| STE buffert | 400 mM sackaros, 50 mM Tris pH 7,8, 20 mM EDTA pH 8,0, 0,6% (vikt / volym) polyvinylpyrrolidon (PVP), 0,2% (vikt / volym) bovint serumalbumin (BSA), O.1% (volym / volym) p-merkaptoetanol (BME) | Buffertblandning som innehåller endast sackaros, Tris och EDTA kan göras upp till en månad i förväg och hållas vid 4 ° C. PVP, BSA och BME bör tillsättas färskt till en alikvot av den erforderliga buffertmängden precis före användning. | Metod nr 1 |
| ST buffert | 400 mM sackaros, 50 mM Tris pH 7,8, 0,6% (vikt / volym) polyvinylpyrrolidon (PVP), 0,1% (vikt / volym) bovint serumalbumin (BSA) | Buffertblandning innehållande endast sackaros och Tris kan göras upp till en månad i förväg och hållas vid 4 ° C. Observera att ST-bufferten inte innehåller EDTA eller BME och innehåller en lägre koncentration av BSA. | Metod nr 1 |
| DNase lager | 2 mg / ml DNas i 0,15 M NaCl till en stamkoncentration av 2 mg / ml | Förvara 200 μl alikvoter vid -20 ° C. För att förbereda DNase-arbetslösning (200 μl DNase-lösning per prov) seTabell 1 nedan. Se fullständiga protokollet nedan för fullständiga detaljer om DNase-digestion. DNase-arbetslösning ska beredas färskt. För att stoppa DNasreaktionen krävs en lösning på 400 mM EDTA pH 8,0 (slutkoncentration som krävs för att stoppa reaktionen är 0,2 M EDTA, se fullständigt protokoll för detaljer). | Metod nr 1 |
| DNase arbetslösning | 0,25 mg / ml DNas och 20 mM MgCl2 i ST Buffert | Förbered fräsch, 200 μl per prov. Koncentrationer som visas är för slutlig reaktionsvolym, så blanda: 62,5 | il 2 mg / ml DNas (baserat på slutlig 500 | il reaktionsvolym), 4 | il 1 M MgCl 2 (baserat på 200 | il DNas lösningsvolymen), och 133.5 pl av ST-buffert för En slutlig volym av 200 pl. | Metod nr 1 |
| Lysisbuffert | 20 mM EDTA pH 8,0; 10 mM Tris pH 7,9; 500 mM guanidin-HCl; 200 mM NaCl; 1% Triton X-100; 0,5 mg / ml lyseringsenzymer frånTrichoderma harzianum | Blanda alla ingredienser utom lyseringsenzymer och förvara vid rumstemperatur. Lyseringsenzymer bör tillsättas färskt till en liten alikvot för omedelbar användning. | Metod nr 2 |
Tabell 1: Recept av hemlagad buffert och arbetslager.
| Koncentrationsarbete | |||||||
| Provnamn | Tom Enhetsvikt (g) | Vikt av fylld enhet (g) | Fylld volym (ul, fylld minus tomma vikter) | Vikt efter 1: a spinnet (20 min *, g) | Volym efter 1: a Spin (ul, fylldMinus tomma vikter) | Vikt efter 2: a Spin (X min *, g) | Volym efter 2: a Spin (ul, fylld minus tomma vikter) |
| Observera att den faktiska återhämtade volymen kommer att vara några ul mindre än beräknad volym. | |||||||
Tabell 2: Koncentrationsarbete.
| namn | Genomspecificitet | Gen-sekvenskälla | Sekvens (5 '- 3') |
| Ta_ACTIN - F | Kärn | Gramene Stillbild IWGSC_CSS_1AS_scaff_3272162: 10,663-12,557 | CAGGTATCGCTGACCGTATGA |
| Ta_ACTIN - R | Kärn | Samma som ovan | GAAGGTAGGGCTGAACAAGAAAC |
| Ta_NAD3 - F | mitochondrial | NCBI-anslutning EU534409.1 | GGTGATGCCAGAAGTCGTTT |
| Ta_NAD3 - R | mitochondrial | Samma som ovan | CAGATCAATCTTGTTAGGAGGTACTG |
| Ta_PSBB - F | kloroplast | NCBI-anslutning KJ592713.1 | GCTACCTTTGCTTTGCTCTTCT |
| Ta_PSBB - R | kloroplast | Samma som ovan | GCTGCCTGTTTCCTTGTAGTT |
Tabell 3: Förteckning över qPCR Primers.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Hittills är de flesta organellära sekvenseringsstudier centrerad på traditionella DC-metoder för att berika för specifikt DNA. Metoder för att isolera organeller från olika växter har beskrivits, inklusive moss 40 ; Monokoter såsom vete 15 och havre 11 ; Och dikotes som arabidopsis 11 , solros 17 och rapsfrö 14 . De flesta protokoll fokuserar på bladvävnad 13 , 14 , 15 , 16 , 17 , varvid vissa har anpassats för en mängd olika vävnadstyper, inklusive frön 11 . Isoleringen av organeller från protoplaster har också visats 41 . Detta är dock inte mottagligt för alla system, och det är inte heller möjligt när vävnaden av intresse är begränsad. Många av dessa orgaNella isoleringsmetoder utformades för att återvinna intakta organeller för specifika experiment, såsom fysiologiska studier. Dessa protokoll är besvärliga och kräver typiskt användning av densitetsgradienter, såsom sackaros- eller Percoll-gradienter, vilka är mycket effektiva vid isolering av specifika organellära fraktioner, men kräver en stor vävnadsinmatning ( dvs. över 5 g och uppåt i kilo beroende på Vävnadstypen). DC-metoden kan emellertid optimeras för att berika för specifika cellulära fraktioner, såsom mitokondrier eller kloroplast, genom att ändra snurrhastigheter och densitetsgradienter. MF-tillvägagångssättet kräver däremot mycket mindre utgångsmaterial (20 mg), men mitokondriella och plastid-DNA kommer att finnas närvarande per deras relativa överflöd i vävnaden som används för DNA-extraktion. Icke desto mindre erbjuder MF-protokollet ett alternativt tillvägagångssätt för isolering av blandat organellärt DNA och är särskilt fördelaktigt för att börja med små mängder vävnad.
T O utvärdera provrenhet efter organellisolering, de flesta studierna hittills använder endast slutpunkts-PCR och gelelektrofores 11 , 12 . Detta ger en rättvis kvalitativ åtgärd av provrenhet. Låga nivåer av amplifiering kan emellertid inte visualiseras på en agarosgel. Få rapporter innehåller mer kvantitativa åtgärder för kvalitetskontroll, som qPCR 14 . För en kvantitativ bedömning av DNA-provrenhet isolerad från båda metoderna utnyttjade vi qPCR och sekvensering för att bestämma hur mycket kärn-DNA kvarstår i provet, såväl som de relativa proportionerna av mitokondriellt mot kloroplast-DNA. Båda metoderna som utvärderas här är effektiva vid avlägsnande av nukleärt DNA. Båda metoderna ger en blandning av mitokondriellt och kloroplast-DNA, om än i olika proportioner.
Växande växter i mörkret (etiolation) rapporteras bidra till att underlätta organellärisolering på grund av minskning av fenolRef "> 13. Men i denna jämförelse fann vi inte någon märkbar fördel att arbeta med etiolerat vävnad över lätta vuxna prover. Även om andelen specialiserade kloroplaster sannolikt kommer att vara högre vid ljusvikt, kommer det totala plastidantalet, som Reflekterad i andelen av läsning av kartläggning till kloroplastgenomet, är oförändrad under olika ljusförhållanden. Därför rekommenderar vi för genomströmsfunktionella analyser, såsom bedömning av heteroplasm i olika vävnader eller under olika stressorer eller för expressionsanalyser, att genomisk sekvensering utförs på Växter odlade under fysiologiskt relevanta förhållanden.
För applikation med kortläsning av sekvenseringsteknologier ger båda teknikerna jämfört här tillräcklig DNA-kvantitet och kvalitet. För att uppnå långa avläsningar av> 20 kb för enkelmolekylsekvenseringsapplikationer är emellertid en större mängd DNA av högre kvalitet nödvändigt. Till exempel helst> 1 μg ren orgaNellärvete-DNA med en molekylvikt> 20 kb är nödvändig för in-house, low-input-protokoll för 20-kb-inmatningsbibliotekspreparat 42 . Nya användarutvecklade protokoll med låg ingång kan minska DNA-kraven ( dvs. till 50 ng eller ännu mindre 20 ), men utmaningen återstår att ha högkvalitativt DNA med hög molekylvikt som går in i bibliotekets preparat. Det är väsentligt att en majoritet av DNA: n är> 20 kb, eftersom mindre fragment kommer att införas företrädesvis i SMRTbell och kasta bort storlekens fördelning av biblioteket 43 . Vi försökte ett antal hemlagade DNA-extraktionsprotokoll och ett antal kommersiella protokoll för DNA-extraktion (ej visad). För vävnadsbladvävnad erhölls det bästa balansen mellan DNA-kvantitet och kvalitet, särskilt längd, med användning av ett kommersiellt kit 27 , 29 . Beroende på växtarter och vävnad av intresse, alternatiVe extraktionsprotokoll kan vara lika passande eller mer fruktbart. Icke desto mindre drar vi slutsatsen att den totala extraktionen av genomisk DNA med hög molekylvikt> 50 kb i följd, följt av fraktionering med MBD2-Fc pulldown-tillvägagångssättet 28 , är mottaglig för långlästsekvensering från begränsat utgångsmaterial. Framtida arbete bör testa gränserna för utgångsmaterialet som krävs efter fraktionering för långtinsättning av bibliotekets förberedelse och efterföljande långlästsekvensering. Kritiskt kan detta tillvägagångssätt tillhandahålla en robust metod för att isolera DNA från ett prov av ett enda blad som är lämpligt för långlästsekvensering, utan helgenomförstärkning. Vi förutser att detta tillvägagångssätt lätt kan anpassas till ytterligare vävnadstyper och i stor utsträckning tillämpas på andra växtarter. Det kommer att vara särskilt användbart i situationer där vävnadsmängderna är begränsande, såsom sekvensering vid enskilda generationer i ett korsningsschema eller i sämre vävnadstyper.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Författarna förklarar att de inte har några konkurrerande intressen.
Nämnandet av handelsnamn eller kommersiella produkter i denna publikation är enbart för att ge specifik information och innebär inte rekommendation eller godkännande av US Department of Agriculture. USDA är en jämställdhetsleverantör och arbetsgivare.
Acknowledgments
Vi skulle vilja erkänna finansiering från Förenta staternas avdelningen för jordbruk och jordbruksforskning och från National Science Foundation (IOS 1025881 och IOS 1361554). Vi tackar R. Caspers för växthusunderhåll och växtvård. Vi tackar också Universitetet i Minnesota Genomics Center, där Illumina biblioteket förberedelser och sekvensering utfördes. Vi är också tacksamma för kommentarerna från tidningsredaktörerna och fyra anonyma granskare som ytterligare förstärkt vårt manuskript. Vi tackar också OECD för ett stipendium till SK för att integrera dessa protokoll för samarbetsprojekt med kollegor i Japan.
Materials
| Name | Company | Catalog Number | Comments |
| 2-mercaptoethanol (beta-mercaptoethanol; BME) | Sigma Aldrich | M3148-100ml | |
| 2-propanol (Isopropyl alcohol/isopropanol), bioreagent | Sigma Aldrich | I9516 | |
| agarose, Bio-Rad Cetified Megabase agarose | Bio-Rad | 1613108 | |
| analytical balance | Mettler Toledo | AB54-S | |
| balance | Mettler Toledo | PB1502-S | |
| bovine serum albumin (BSA) | Sigma Aldrich | B4287-25G | |
| Ceramic grinding cylinders, 3/8in x 7/8in | SPEX SamplePrep | 2183 | |
| Cryogenic Blocks compatible with tissue homogenizer for holding 50 mL tubes | SPEX SamplePrep | 2664 | |
| DNaseI | Sigma | DN25 | |
| ethanol, absolute | Decon Laboratories | 2716 | |
| Ethylenediamine Tetraacetic Acid (EDTA), 0.5 M Solution, pH 8.0 | Fisher | BP2482-500 | |
| gel imaging system | |||
| gel stain | Such as GelRed or Ethidium Bromide | ||
| grinding pestle, wide tip for 2 mL conical tubes | |||
| Guanidine-HCl, 8 M solution | ThermoFisher | 24115 | |
| LightCycler 480 SYBR Green I Master | Roche | 4707516001 | |
| liquid nitrogen | |||
| Lysing enzymes from Trichoderma harzianum | Sigma | L1412 | |
| Magnesium Chloride | G Bioscience | 24115 | |
| magnetic rack | ThermoFisher | A13346 | |
| microcentrifuge tubes, LoBind 1.5 mL | Eppendorf | 22431021 | |
| microcentrifuge tubes, standard nuclease-free 1.5 mL | Eppendorf | ||
| microcentrifuge, refrigerated | Sorvall | Legend X1R | Or equivalent product, must be capable of reaching at least 18,000 x g with rotors for 50 mL tubes, Oak Ridge tubes, and 1.5 mL tubes |
| microcentrifuge, room temperature | Eppendorf | 5424 | Or equivalent product, must be capable of reaching at least 18,000 x g with rotor for 1.5 mL and 2 mL microcentrifuge tubes |
| Microcon DNA Fast Flow Centrifugal Filter Units | EMD Millipore | MRCFOR100 | |
| Miracloth, 1 square per sample cut to fit funnel | EMD Millipore | 475855 | |
| NEBNext Microbiome DNA Enrichment Kit | New England Biolabs | E2612L | |
| parafilm | Parafilm M | PM992 | |
| plastic pots and trays | |||
| polyvinylpyrrolidone (PVP) | Fisher | BP431-100 | |
| Proteinase K | Qiagen | 19131 | |
| Pulsed-Field Gel Electrophoresis rig (e.g. CHEF DR III) | Bio-Rad | 1703697 | |
| purification beads, Agencourt AMpureXP beads | Beckman Coulter | A63881 | |
| QIAamp DNA Mini Kit | Qiagen | 51304 | |
| Qiagen 20/g Genomic Tip DNA Extraction Kit | Qiagen | 10223 | |
| Qiagen Buffer EB (elution buffer) | Qiagen | 19086 | |
| Qiagen DNA Extraction Buffer Set | Qiagen | 19060 | |
| QiaRack | Qiagen | 19015 | |
| qPCR machine (e.g. Roche Light Cycler 480) | Roche | ||
| qPCR plate sealing film | Roche | 4729757001 | |
| qPCR plate, 96 well plate | Roche | 4729692001 | |
| Qubit assay tubes | Life Technologies | Q32856 | |
| Qubit Broad Spectrum assay kit | Life Technologies | Q32850 | |
| Qubit High Sensitivity assay kit | Life Technologies | Q32851 | |
| RNaseA | Qiagen | 19101 | |
| Serological pipettes (20 mL) and pipet-aid | Fisher | 13-678-11 | |
| Small funnels, 1 per sample | |||
| Sodium Chloride | Ambion | AM9759 | |
| Soft paintbrush, 2 per sample | |||
| SPEX SamplePrep 2010 Geno/Grinder or another type of tissue homogenizer | SPEX SamplePrep | Or another comparable tissue homogenizer. If you do not have access to a tissue homogenizer, then grinding in a pre-chilled mortar and pestle will suffice (see protocol for details). However, a homogenizer will give more consistent results and total homogenization time is reduced. | |
| Sucrose | Omnipure | 8550 | |
| TBE | |||
| thermomixer | |||
| Tris | Sigma | T2819-100ml | |
| Triton X-100 | Promega | H5142 | |
| tube rotater | |||
| tubes, 50 mL conical polypropylene | Corning | 352070 | |
| tubes, 50 mL high-speed polypropylene | ThermoScientific/Nalgene | 3119-0050 | e.g. Nalgene Oakridge tubes or equivalent |
| vermiculite | |||
| water bath | |||
| water, sterile and certified Nuclease-free | Fisher | 1481 | |
| water, sterile milliQ |
References
- Liberatore, K. L., Dukowic-Schulze, S., Miller, M. E., Chen, C., Kianian, S. F. The role of mitochondria in plant development and stress tolerance. Free Radic Biol Med. 100, 238-256 (2016).
- Samaniego Castruita, J. A., Zepeda Mendoza, M. L., Barnett, R., Wales, N., Gilbert, M. T. Odintifier--A computational method for identifying insertions of organellar origin from modern and ancient high-throughput sequencing data based on haplotype phasing. BMC Bioinformatics. 16 (232), 1-13 (2015).
- Zhang, T., Zhang, X., Hu, S., Yu, J. An efficient procedure for plant organellar genome assembly, based on whole genome data from the 454 GS FLX sequencing platform. Plant Methods. 7 (38), 1-8 (2011).
- Wambugu, P. W., Brozynska, M., Furtado, A., Waters, D. L., Henry, R. J. Relationships of wild and domesticated rices (Oryza AA genome species) based upon whole chloroplast genome sequences. Sci Rep. 5 (13957), 1-9 (2015).
- Iorizzo, M., et al. De novo assembly of the carrot mitochondrial genome using next generation sequencing of whole genomic DNA provides first evidence of DNA transfer into an angiosperm plastid genome. BMC Plant Biol. 12 (61), 1-17 (2012).
- Park, S., et al. Complete sequences of organelle genomes from the medicinal plant Rhazya stricta (Apocynaceae) and contrasting patterns of mitochondrial genome evolution across asterids. BMC Genomics. 15 (405), 1-18 (2014).
- Skippington, E., Barkman, T. J., Rice, D. W., Palmer, J. D. Miniaturized mitogenome of the parasitic plant Viscum scurruloideum is extremely divergent and dynamic and has lost all nad genes. Proc Natl Acad Sci U S A. 112 (27), E3515-E3524 (2015).
- Wicke, S., Schneeweiss, G. M. Chapter 1. Next Generation Sequencing in Plant Systematics. Hörandl, E., Appelhans, M. , Koeltz Scientific Books. (2015).
- Sloan, D. B. One ring to rule them all? Genome sequencing provides new insights into the 'master circle' model of plant mitochondrial DNA structure. New Phytol. 200 (4), 978-985 (2013).
- Woloszynska, M. Heteroplasmy and stoichiometric complexity of plant mitochondrial genomes--though this be madness, yet there's method in't. J Exp Bot. 61 (3), 657-671 (2010).
- Ahmed, Z., Fu, Y. B. An improved method with a wider applicability to isolate plant mitochondria for mtDNA extraction. Plant Methods. 11 (56), 1-11 (2015).
- Ejaz, M., et al. Comparison of small scale methods for the rapid and efficient extraction of mitochondrial DNA from wheat crop suitable for down-stream processes. Genet Mol Res. 13 (4), 10320-10331 (2014).
- Eubel, H., Heazlewood, J. L., Millar, A. H. Isolation and subfractionation of plant mitochondria for proteomic analysis. Methods Mol Biol. 355, 49-62 (2007).
- Hao, W., Fan, S., Hua, W., Wang, H. Effective extraction and assembly methods for simultaneously obtaining plastid and mitochondrial genomes. PLoS One. 9 (9), e108291 (2014).
- Pomeroy, M. K. Studies on the respiratory properties of mitochondria isolated from developing winter wheat seedlings. Plant Physiol. 53 (4), 653-657 (1974).
- Taylor, N. L., Stroher, E., Millar, A. H. Arabidopsis organelle isolation and characterization. Methods Mol Biol. 1062, 551-572 (2014).
- Triboush, S. O., Danilenko, N. G., Davydenko, O. G. A method for isolation of chloroplast DNA and mitochondrial DNA from Sunflower. Plant Mol Biol Rep. 16 (2), 183-189 (1998).
- Pinard, R., et al. Assessment of whole genome amplification-induced bias through high-throughput, massively parallel whole genome sequencing. BMC Genomics. 7 (216), 1-21 (2006).
- Lamble, S., et al. Improved workflows for high throughput library preparation using the transposome-based Nextera system. BMC Biotechnol. 13 (104), 1-10 (2013).
- Raley, C., et al. Preparation of next-generation DNA sequencing libraries from ultra-low amounts of input DNA: Application to single-molecule, real-time (SMRT) sequencing on the Pacific Biosciences RS II. bioRxiv. , (2014).
- Tsai, Y. C., et al. Resolving the Complexity of Human Skin Metagenomes Using Single-Molecule Sequencing. MBio. 7 (1), e01948 (2016).
- Feehery, G. R., et al. A method for selectively enriching microbial DNA from contaminating vertebrate host DNA. PLoS One. 8 (10), e76096 (2013).
- Yigit, E., Hernandez, D. I., Trujillo, J. T., Dimalanta, E., Bailey, C. D. Genome and metagenome sequencing: Using the human methyl-binding domain to partition genomic DNA derived from plant tissues. Appl Plant Sci. 2 (11), 1-6 (2014).
- Noyszewski, A. K., et al. Accelerated evolution of the mitochondrial genome in an alloplasmic line of durum wheat. BMC Genomics. 15 (67), 1-16 (2014).
- Qiagen. QIAamp DNA Mini and Blood Mini Handbook. , 5th ed, Available from: https://www.qiagen.com/ch/resources/ (2016).
- E.M. Corporation. User Guide: Microcon Centrifugal Filter Devices. , Available from: http://www.emdmillipore.com/US/en/product/Microcon-DNA-Fast-Flow-Centrifugal-Filter-Unit-with-Ultracel-membrane,MM_NF-MRCF0R100 (2013).
- Qiagen. User developed protocol: Isolation of genomic DNA from plants and filamentous fungi using the QIAGEN Genomic-tip - (EN). , Available from: https://www.qiagen.com/ch/resources/ (2001).
- New England BioLabs, Inc.. NEBNext Microbiome DNA Enrichment Kit: Instruction Manual Version 4.0. , Available from: http://www.neb.com/~/media/Catalog/All-Products/371BCB5A557C462D95D1E45E15BBFEA3/Datacards or Manuals/E2612Manual.pdf (2015).
- Qiagen. QIAGEN Genomic DNA Handbook. , Available from: https://www.qiagen.com/ch/resources/ (2012).
- PacificBiosciences. Guidelines for Using the BIO-RAD® CHEF Mapper® XA Pulsed Field Electrophoresis System. , Available from: http://www.pacb.com/wp-content/uploads/Unsupported-Guidelines-Using-BIO-RAD-CHEFMapper-XA-Pulsed-Field-Electrophoresis.pdf (2016).
- Andrews, S. FastQC: A quality control tool for high throughput sequence data. , Available from: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (2016).
- Bolger, A. M., Lohse, M., Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 30 (15), 2114-2120 (2014).
- Ogihara, Y., et al. Structural dynamics of cereal mitochondrial genomes as revealed by complete nucleotide sequencing of the wheat mitochondrial genome. Nucleic Acids Res. 33 (19), 6235-6250 (2005).
- Ogihara, Y., et al. Structural features of a wheat plastome as revealed by complete sequencing of chloroplast DNA. Mol Genet Genomics. 266 (5), 740-746 (2002).
- International Wheat Genome Sequencing Consortium (IWGSC). A chromosome-based draft sequence of the hexaploid bread wheat (Triticum aestivum) genome. Science. 345 (6194), (2014).
- Langmead, B., Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat Methods. 9 (4), 357-359 (2012).
- Bendich, A. J. Why do chloroplasts and mitochondria contain so many copies of their genome? Bioessays. 6 (6), 279-282 (1987).
- Kumar, R. A., Oldenburg, D. J., Bendich, A. J. Changes in DNA damage, molecular integrity, and copy number for plastid DNA and mitochondrial DNA during maize development. J Exp Bot. 65 (22), 6425-6439 (2014).
- Ma, J., Li, X. Q. Organellar genome copy number variation and integrity during moderate maturation of roots and leaves of maize seedlings. Curr Genet. 61 (4), 591-600 (2015).
- Lang, E. G., et al. Simultaneous isolation of pure and intact chloroplasts and mitochondria from moss as the basis for sub-cellular proteomics. Plant Cell Rep. 30 (2), 205-215 (2011).
- Tobin, A. K. Subcellular fractionation of plant tissues. Isolation of chloroplasts and mitochondria from leaves. Methods Mol Biol. 59, 57-68 (1996).
- PacificBiosciences. Procedure & Checklist - 10 kb to 20 kb Template Preparation and Sequencing with Low (100 ng) Input DNA. , Available from: http://www.pacb.com/wp-content/uploads/Procedure-Checklist-10-20kb-Template-Preparation-and-Sequencing-with-Low-Input-DNA.pdf (2015).
- PacificBiosciences. Template Preparation and Sequencing Guide. , Available from: http://www.pacb.com/wp-content/uploads/2015/09/Guide-Pacific-Biosciences-Template-Preparation-and-Sequencing.pdf (2014).
