

Summary
Sammenligningen og optimaliseringen av to organellar DNA-anrikningsmetoder presenteres: Tradisjonell differensial sentrifugering og fraksjonering av totalt gDNA basert på metyleringsstatus. Vi vurderer den resulterende DNA-kvantiteten og kvaliteten, demonstrerer ytelse i kortlestende neste generasjons sekvensering, og drøfter potensialet for bruk i langlestende enkeltmolekylesekvensering.
Abstract
Plantorganellgenomer inneholder store, repeterende elementer som kan gjennomgå parring eller rekombinasjon for å danne komplekse strukturer og / eller sub-genomiske fragmenter. Organellargener finnes også i blandinger i en gitt celle eller vevstype (heteroplasm), og en overflod av subtyper kan endres gjennom utvikling eller når det er under stress (substøkiometrisk forskyvning). Next-generation sequencing (NGS) teknologier er nødvendig for å få dypere forståelse av organellar genom struktur og funksjon. Tradisjonelle sekvenseringsstudier bruker flere metoder for å oppnå organellært DNA: (1) Hvis en stor mengde startvev anvendes, blir den homogenisert og underkastet differensiell sentrifugering og / eller gradientrensing. (2) Hvis en mindre mengde vev er brukt ( dvs. hvis frø, materiale eller rom er begrenset), utføres samme prosess som i (1), etterfulgt av hel-genom-amplifisering for å oppnå tilstrekkelig DNA. (3) Bioinformatikkanalyse kan brukes til å seqUence det totale genomiske DNA og å analysere organellar leser. Alle disse metodene har iboende utfordringer og avvik. I (1) kan det være vanskelig å oppnå en så stor mengde startvev; I (2) kunne helgenom forsterkning innføre en sekvenseringsforspenning; Og i (3) kan homologi mellom atom- og organellargener forstyrre montering og analyse. I planter med store atomgener er det fordelaktig å berike for organellært DNA for å redusere sekvenseringskostnader og sekvenskompleksitet for bioinformatikkanalyser. Her sammenligner vi en tradisjonell differensial sentrifugeringsmetode med en fjerde metode, en tilpasset CpG-metyl-nedtrekks-tilnærming, for å separere det totale genomiske DNA i kjernefysiske og organelle fraksjoner. Begge metodene gir tilstrekkelig DNA for NGS, DNA som er høyt beriket for organellære sekvenser, om enn i forskjellige forhold i mitokondrier og kloroplaster. Vi presenterer optimaliseringen av disse metodene for hvetebladvev og diskuterer store fordeler og dUlemper ved hver tilnærming i sammenheng med prøveinngang, protokolllette og nedstrømsapplikasjon.
Introduction
Genomsekvensering er et kraftig verktøy for å dissekere det underliggende genetiske grunnlaget for viktige plantegenskaper. De fleste genomsekventeringsstudier fokuserer på atomgenometinninnholdet, da flertallet av gener ligger i kjernen. Organellargener, inkludert mitokondriene (på tvers av eukaryoter) og plastider (i planter, spesialisert form, kloroplast, arbeider i fotosyntese) bidrar imidlertid til betydelig genetisk informasjon som er avgjørende for organisasjonsutvikling, stressrespons og total kondisjon 1 . Organellargener er typisk inkludert i totale DNA-ekstrakter beregnet for nukleär genom-sekvensering, selv om metoder for å redusere organelle tall før DNA-ekstraksjon også benyttes 2 . Mange studier har brukt sekvenseringsresultater fra totale gDNA ekstraksjoner for å samle organellar genomene 3 , 4 , 5 ,Xref "> 6 , 7. Men når målet for studien er å fokusere på organellargener, øker bruk av total gDNA sekvenseringskostnadene fordi mange leser er" tapt "til kjernefysiske DNA-sekvensene, særlig i planter med store atomgener . Dessuten, på grunn av duplisering og overføring av organellar sekvenser i det nukleære genom og mellom organeller, løse den riktige tilordning stilling av sekvense leser til den riktige genomet er bioinformatically utfordrende 2, 8. rensingen av organellar genomer fra den nukleære genom er ett Strategi for å redusere disse problemene. Ytterligere bioinformatikk strategier kan brukes til å skille leser det kartet til regioner av homologi mellom mitokondriene og kloroplaster.
Mens organellargenene fra mange plantearter er blitt sekvensert, er lite kjent om bredden av organellargenoms mangfoldTilgjengelig i villpopulasjoner eller i dyrkede avlspuljer. Organellærgener er også kjent for å være dynamiske molekyler som gjennomgår betydelig strukturell omlegging på grunn av rekombinasjon mellom gjentasekvenser 9 . Videre er flere kopier av organellargenomet inneholdt i hver organell, og flere organeller er inneholdt i hver celle. Ikke alle kopier av disse genomene er identiske, som er kjent som heteroplasmisk. I motsetning til det kanoniske bildet av "master sirkler", er det nå voksende bevis for et mer komplekst bilde av organellar genom strukturer, inkludert sub-genomiske sirkler, lineære kromosomer, lineære concatamers og forgrenede strukturer 10 . Samlingen av planteorganellgener er ytterligere komplisert av deres forholdsvis store størrelser og betydelige inverterte og direkte gjentagelser.
Tradisjonelle protokoller for organellær isolasjon, DNA rensing og etterfølgende gen E-sekvensering er ofte besværlig og krever store mengder vevsinngang, med flere gram oppover på hundrevis av gram ungt bladvev som er nødvendig som utgangspunkt 11 , 12 , 13 , 14 , 15 , 16 , 17 . Dette gjør organellargenomsekvensering utilgjengelig når vev er begrenset. I noen situasjoner er frømengder begrenset, for eksempel når det er nødvendig å sekvensere på generasjonsbasis eller i mannlige sterile linjer som må opprettholdes ved kryssing. I disse situasjonene kan organellar-DNA bli renset og deretter utsatt for hel-genom-amplifisering. Imidlertid kan helgenomforsterkning introdusere signifikant sekvenseringsforspenning, hvilket er et spesielt problem når man vurderer strukturell variasjon, sub-genomiske strukturer og heteroplasminnivåer> 18. Nylige fremskritt i biblioteket forberedelse for kortleste sekvensering teknologier har overvinne lav-inngangsbarrierer for å unngå hel-genom forsterkning. For eksempel tillater Illumina Nextera XT-bibliotekets forberedelsessett så lite som 1 ng DNA som skal brukes som inngang 19 . Standardbibliotekspreparasjoner for langleste sekvenseringsapplikasjoner, som for eksempel PacBio eller Oxford Nanopore-sekvenseringsteknologi, krever imidlertid fortsatt en relativt høy mengde inngangsd DNA, som kan utgjøre en utfordring for organellær genom-sekvensering. Nylig er nye, brukervennlige, langleste sekvenseringsprotokoller blitt utviklet for å redusere inngangsmengder og for å bidra til å fremme genom-sekvensering i prøver hvor det er vanskelig å få mikrogram-mengder av DNA 20 , 21 . Imidlertid er oppnåelse av høymolekylære, rene organellære fraksjoner for å matche inn i disse bibliotekspreparatene en utfordring.
Vi søkte tO sammenligne og optimalisere organellar DNA-anriknings- og isoleringsmetoder egnet for NGS uten behov for helgenomdannelse. Spesielt var målet vårt å bestemme beste praksis for å berike for organellært DNA med høy molekylvekt fra begrensede utgangsmaterialer, for eksempel en undersampling av et blad. Dette arbeidet presenterer en komparativ analyse av metoder for å berike for organellært DNA: (1) en modifisert, tradisjonell differensialsentrifugeringsprotokoll versus (2) en DNA-fraksjonsprotokoll basert på bruk av et kommersielt tilgjengelig DNA-CpG-metylbindende domeneprotein-nedtrekks-tilnærming 22 påført plantevev 23 . Vi anbefaler beste praksis for isolering av organellar DNA fra hveteblad vev, som lett kan utvides til andre planter og vevstyper.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. Generering av plantematerialer for organellarisolasjon og DNA-ekstraksjon
- Standard vekst av hveteplanter
- Plante frø i vermikulitt i små, firkantede potter med 4 - 6 frø per hjørne. Overfør til et drivhus eller vekstkammer med 16 h lyssyklus, 23 ºC dag / 18 ºC natt.
- Vann plantene hver dag. Gjød plantene med ¼ ts granulær 20-20-20 NPK gjødsel ved spiring og 7 dager etter spiring.
- Alternativ etiolering av hveteplanter
- Følg trinn 1.1, men legg potene i et mørkt vekstkammer, 23 ° C i 16 timer / 18 ºC i 8 timer. Alternativt, dekke plantene i drivhuset ( f.eks. Med en lagringsbeholder, men riktig ventilasjon må opprettholdes).
- Vekst og vevsinnsamling
- Vok plantene i 12-14 dager. For de fleste hvete genotypeS, 75 - 100 frøplanter gir rundt 10-12 g vev, som er tilstrekkelig for to organellar ekstraksjoner ved hjelp av differensial sentrifugeringsmetoden (avsnitt 2); Bare en plante er nødvendig dersom man bruker DNA CpG-metyleringsbasert nedtrekksmetode til fraksjonering av organellar fra nukleært DNA (avsnitt 3).
- Hvis du bruker differensial sentrifugering tilnærming, samle vev frisk og fortsett med det samme for å behandle prøvene, som beskrevet i avsnitt 2.
- Hvis du bruker CpG-methyl-nedtrekksmetoden, høste 20 mg seksjoner av ungt bladvev inn i mikrocentrifugerør (bruk enten standardvokset eller etiolert vev, se representativresultater ). Snapshot på flytende nitrogen og frys ved -80 ºC til bruk. Fortsett til pulldown fraksjonering av DNA, som beskrevet i avsnitt 3.
2. Metode nr. 1: DNA-ekstraksjon ved bruk av differensiell sentrifugering (DC)
MERK: diffErential sentrifugeringsprotokoll ble modifisert fra to publikasjoner som optimaliserte forhold for å isolere begge organeller, men berike for mitokondrier 17 , 24 . Den resulterende protokollen er mindre tidsintensiv og bruker færre giftige kjemikalier enn de tidligere metodene. Spesielt gjorde vi endringer i buffere og vaske trinn, inkludert tilsetning av polyvinylpyrrolidon (PVP) til STE-ekstraksjonsbufferen og eliminering av det endelige vaske-trinnet i NETF-buffer, som inneholder natriumfluorid (NaF).
Forsiktig: Forberedelse og bruk av STE-buffer skal utføres under kjemisk avtrekksdeksel med riktig personlig verneutstyr, da denne buffer inneholder 2-merkaptoetanol (BME).
- Ting å gjøre før du starter
- Sørg for at alt utstyr er ekstremt rent, og autoklaver alt utstyr som kan autoklaveres ( f.eks. Slipesylindere, høyhastighets sentriFuge rør, etc. ).
MERK: Filtertips anbefales for alle trinn som krever pipettering for å unngå krysskontaminering. - Se listen over nødvendige utstyr og reagenser og lag de nødvendige buffere og arbeidslager for metode nr. 1 ( tabell 1 ). Kyll de kryogene slipeblokkene til -20 ºC og rotorene og bufferne til 4 ºC, sett mikrocentrifugen til 4 ºC, og slå på et 37 ºC vannbad.
- Sørg for at alt utstyr er ekstremt rent, og autoklaver alt utstyr som kan autoklaveres ( f.eks. Slipesylindere, høyhastighets sentriFuge rør, etc. ).
- Isolering av organeller
- Høst 5 g ferskt vev og skyll det i kaldt, sterilt vann i et kjølt beger på is.
MERK: Hold alltid prøver på is under alle operasjoner og transporter til og fra sentrifuger, avtrekksvifter etc. Alternativt kan du arbeide i et kaldrom hvis det er tilgang til tilstrekkelig plass og utstyr for å utføre protokollen. - Ved hjelp av sakse, kutte bladvev i ~ 1 cm-stykker direkte inn i et 50 ml rør som inneholder to keramisk slipingsylindere.
MERK: Rengjør eller bytt saks mellom prøver for å unngå krysskontaminering. - Hvis det ikke er vevshomogenisator, bruk en mørtel og pestel og følg for å erstatte trinn 2.2.4 - 2.2.9.
- Klipp bladvevet til en forkjølt mørtel på is. Grind prøvene i 2 - 3 min i 15 ml STE (i avtrekksdekselet).
- Hell bufferen (la vevet stå i mørtelet) gjennom en trakt som inneholder ett lag av for-våt, steril filtreringsklut (~ 22 til 25 μm porestørrelse, se hovedprotokollen for detaljer) i et annet 50 ml rør . Tilsett ytterligere 10 ml STE til mørtel og pestle og homogeniser igjen.
- Hell det homogeniserte vevet og bufferen i samme trakten. Skyll morter og pestle med 10 ml STE og hell den inn i trakten. Klem og vri ut filtrertøyet i trakten for å gjenopprette så mye væske som mulig.
MERK: Bytt hansker mellom prøver for å unngå krysskontaminering. Fortsett med proffTocol i trinn 2.2.10.
- Tilsett 20 ml STE (i avtrekksdekselet) til hvert 50 ml rør.
- Plasser prøvene i forkjølte kryogene slipeblokker i et vevslipingsapparat og grind prøvene i 2 x 30 s ved 1750 rpm. Vri prøveposisjonene og plasser prøvene på is i ~ 1 min mellom grindene.
MERK: En mørtel og pestle, blender eller annen vev sliping / homogeniseringsanordning kan brukes i dette trinnet. Imidlertid vil hver metode påvirke den resulterende DNA-kvaliteten til forskjellige grader, og derfor bør DNA-lengde og kvalitet vurderes før de fortsetter med nedstrømsapplikasjoner. - Sett en trakt i et rent 50 ml rør plassert i is. Legg ett lag med filtreringsklut i trakten og pre-våt den med 5 ml STE. Ikke kast bort gjennomstrømmingen.
- Hell det homogeniserte vevet i trakten. Skyll slipeslangen med 15 ml STE, sett sammen og vend omrøret for å skylle veggene og lokket og hell inn i funnel.
- Fjern forsiktig de keramiske steinene, og klem deretter og vri ut filtrertøyet i trakten.
MERK: Bytt hansker mellom prøver for å unngå krysskontaminering. - Wrap tube caps med parafilm for å unngå spredning. Sentrifuger ved 2000 xg i 10 minutter ved 4 ºC.
- Forsiktig suge supernatanten ved hjelp av en serologisk pipette (unngå å forstyrre pelleten) og plasser den i et 50 ml høyhastighets sentrifugerør (hvis rørene ikke har tette forseglingspakninger, pakk rørkapslene sammen med parafilm for å unngå spredning). Kast pelletsene.
- Balanse rørene til innenfor 0,1 g ved hjelp av STE og sentrifuger den resulterende supernatanten i 20 minutter ved 18 000 xg og 4 ºC. For å balansere rørene, plasser en liten isbukk på balansen, tørk skalaen og vei prøvene på is for å holde dem kalde. Alternativt, bruk en balanse og en hette i et kaldt rom.
- Kast supernatanten. Tilsett 1 mL ST til pellet og oppsug forsiktig ossLegg en myk pensel. Legg til 24 ml ST (sluttvolum på 25 ml) og bland / virvel ( dvs. trykk på pensel på siden av røret for å fjerne all væske).
- Balanse rørene til innenfor 0,1 g ved å bruke ST. Sentrifuger i 20 minutter ved 18.000 xg og 4 ºC. I mellomtiden forberede DNaseI-oppløsning (se tabell 1 for oppskrifter av lager og arbeidsoppløsning). For hver prøve, lag en 200 μl alikvot i et 1,5 ml rør.
- Kast supernatanten, flekk røret og suspender pelleten (fortsatt i et høyhastighets sentrifugerør) i 300 μL ST ved hjelp av en myk pensel. Plasser penselen i det tidligere preparerte 1,5 ml røret som inneholder 200 μl DNaseI-oppløsning og virv penselbrettet for å fjerne eventuell resterende pellet fast i børsten. Pipetter DNaseI-løsningen tilbake i høyhastighets-sentrifugerøret og rør forsiktig for å blande.
- Inkuber ved 37 ° C i 30 minutter i et vannbad (pakk parafilmen rundt toppen av røret for å forhindre kondens lekkasjeG inn i lokket). Bland forsiktig ved å virvle 2 ganger under inkubasjon.
- Pett forsiktig pelletblandingen ut av røret ved å bruke en pipettespiss med en bred åpning og plasser den i et 1,5 ml lavt rør. Tilsett 500 μL 400 mM EDTA, pH 8,0, til høyhastighets sentrifugerøret og forsiktig pipetten for å få hele restpellet ut av røret. Overfør EDTA til samme 1,5 ml, lavbindingsrør som pelletsblanding og bland forsiktig ved inversjon.
- Sentrifuger ved 18 000 xg i 20 minutter ved 4 ºC. Kast supernatanten, slett røret og bruk umiddelbart for DNA-isolasjon. Hvis det er nødvendig, frys pellets ved -20 ºC, men dette kan resultere i en avkastningsreduksjon, da resterende DNaseI kan nedbryte prøve-DNA dersom det ikke umiddelbart behandles.
- Høst 5 g ferskt vev og skyll det i kaldt, sterilt vann i et kjølt beger på is.
- DNA-ekstraksjon fra isolerte organeller ved hjelp av en kommersiell kolonnebasert tilnærming
MERK: Se kithåndboken for full protokoll 25 , og se nedenfor for endringer. PrÅ overføre direkte fra organellarisolasjon til DNA-ekstraksjon er foretrukket. Gjentatt frysing og tining vil redusere DNA-fragmentstørrelser og føre til DNA-nedbrytning av gjenværende DNaseI. Begrens vortexing eller kraftig pipettering, da dette kan skjære DNA. Bruk av lavbindende mikrocentrifugerør anbefales for å maksimere DNA-gjenvinning.- DNA-ekstraksjonsprosedyre
MERK: Les den detaljerte kommersielle protokollen 25 før du begynner å sikre at bufferne er riktig lagret / lagret og at spin-kolonne prosedyrene blir forstått.- Tilsett 180 μL Buffer ATL direkte inn i røret med pellet (tint hvis det tidligere er frosset og ekvilibrert til romtemperatur på benken).
- Fortsett med trinn 3 i protokollen for "DNA-rensninger fra væsker" i kithåndboken, med følgende modifikasjoner: en 30 min lysis i trinn 3 inkluderer den valgfrie RNase A-fordøyelsen og elueres i 3 x 200 μl AE ( Hver til en seParate tube og deretter kombinere elutions).
- Lagre en alikvot (minst 20 μL) for qPCR (se trinn 4.1). For å kvantifisere før konsentrering, lagre ytterligere 1 μL for høy følsomhetskvantifisering.
- Fortsett med prøvekonsentrasjon hvis ønskelig.
- DNA-ekstraksjonsprosedyre
- Prøvekonsentrasjon med kommersielle filterenheter
MERK: Se den kommersielle protokollen 26 for flere detaljer. Avhengig av nedstrøms bruk kan det ikke være nødvendig å utføre prøvekonsentrasjon ( f.eks. For sluttpunkts-PCR og qPCR-applikasjoner). For NGS-bibliotekskonstruksjon vil det imidlertid sannsynligvis være nødvendig å konsentrere utvunnet organellært DNA oppnådd etter DNA-ekstraksjon.- Konsentrasjonskolonneprosedyre
- Forsiktig forsiktig (se tabell 2 ) den tomme filterenheten (uten et rør) på et rent stykke veiepapir på en digital analytisk balanse. Legg inn vekten.
- PiPette de kombinerte elusjonene inn i filterenheten og veid omhyggelig igjen.
MERK: Den kommersielle håndboken 26 sier at maksimalt volum på filterenheten er 500 μL, men opptil 575 μL kan legges til enheten samtidig uten overløp. - Sett den fylte filterenheten forsiktig inn i et rør (utstyrt med kolonnene). Sentrifuge ved 500 xg for ønsket tid for å oppnå det nødvendige konsentratvolumet. For et prøvevolum på ~ 575 μL vil en 20-minutters spin vanligvis resultere i et konsentratvolum på 15-30 μL.
- Fjern filterenheten fra røret og vei igjen. Bruk tabellen for å bestemme om ønsket konsentratvolum er oppnådd. Hvis ikke, sentrifuger igjen ved 500 xg i kortere tid og veier igjen; Gjenta til ønsket konsentratvolum er nådd.
- Sett et nytt rør (følger med kolonnene) over toppen av filterenheten og inverter. Sentrifuger i 3 minutter ved 1000 xg for å overføre coSentrere til røret.
- Bestem volumet som er gjenopprettet. Dette vil vanligvis være ~ 3 - 5 μL mindre enn det beregnede volumet, på grunn av filterretensjon. Hvis overkonsentrert, fortynn med sterilt vann eller TE for å oppnå ønsket volum.
- Kvantifiser DNA ved å bruke høy følsom kvantifisering (per produsentens instruksjoner).
- Konsentrasjonskolonneprosedyre
3. Metode nr. 2: Metylfraksjonering (MF) tilnærming til berikning for organellært DNA fra totalt genomisk DNA
MERK: Denne protokollen ble modifisert fra en brukerutviklet DNA-ekstraksjonsprotokoll for planter og sopp 27 og den kommersielle Microbiome DNA Enrichment Kit-protokollen 28 . I teorien kan en hvilken som helst DNA-isolasjonsprotokoll som gir DNA med høy molekylvekt, brukes til nedtrekkingen. For kortlestende sekvensering er enhver ekstraksjon som gir overvekt> 15 kb fragmenter tilstrekkelig til bruk i uttrekksdyktigheten. For loNg-lese-sekvensering, kan større fragmenter være ønskelige. Derfor optimaliserte vi denne protokollen for å gi DNA med høy molekylvekt.
- Isolering av totalt DNA
MERK: Se listen over nødvendige utstyr og reagenser og lag de nødvendige buffere og arbeidslager for metode nr. 2 ( tabell 1 ). Tilsett lyseringsenzymer til lysisbuffertmassen for å gjøre lysisbufferens arbeidsoppløsning. Slå på termomixeren og sett den til 37 ° C. Slå på vannbadet til 50 ° C og plasser QF-buffer i badet. Plasser 70% EtOH i fryseren og sett mikrocentrifugen til 4 ° C.- Total DNA-ekstraksjon ved bruk av kommersielle DNA-ekstraksjonskolonner
MERK: Før du starter, les den kommersielle håndboken 29 for detaljert informasjon om bruk av tyngdekraftstrøm anionbytterkolonnene. Kolonnene kan settes opp ved hjelp av en spesialstativ eller plasseres over rørene ved hjelp av de medfølgende plastringene. Alle trinnene, inkludert gEnomic tips, bør få lov til å fortsette med tyngdekraften flyt, og resterende væske bør IKKE bli tvunget gjennom.- Grind 20 mg frosset vev i flytende nitrogen i et 2 ml lavt bindingsrør ved hjelp av håndholdte slipepestler designet for 2 ml rør.
- Tilsett 2 ml lysisbuffer arbeidsløsning (rørene vil være veldig fulle).
- Inkuber i en termomixer ved 37 ° C i 1 time med forsiktig omrøring ved 300 rpm. Hvis en termomixer ikke er tilgjengelig, er det et passende alternativ å inkubere på en varmeblokk og blande ved forsiktig flicking hvert 15. minutt.
- Tilsett 4 μl RNase A (100 mg / ml, sluttkonsentrasjon på 200 μg / mL). Inverter for å blande og inkuber i en termomixer i 30 minutter ved 37 ° C, med forsiktig omrøring ved 300 rpm.
- Tilsett 80 μl proteinase K (20 mg / ml, sluttkonsentrasjon på 0,8 mg / ml), inverter for å blande, og inkuber i en termomixer i 2 timer ved 50 ° C med forsiktig omrøring ved 300 rpm.
- Sentrifuger i 20 minutter ved 4 ° C og 15.000 xg for å pelletere uoppløselig rusk.
- Mens prøvene er sentrifugering, ekvilibrere kolonnene med 1 ml Buffer QBT og la kolonnen tømme ved tyngdekraftstrømmen.
- Bruk en pipettespiss med bred boring for raskt å bruke prøven (unngår pellet) til den ekvilibrerte kolonnen og la den strømme helt gjennom kolonnen. Hvis prøven blir overskyet, filtrer eller centrifuger igjen før påføring i kolonnen (se handelshåndboken for detaljer 29 ).
- Når prøven er fullt inn i harpiksen, vask kolonnen med 4 x 1 ml Buffer QC.
- Suspens kolonnen over et rent, 2 ml, lavt bindende mikrocentrifugerør. Eluk det genomiske DNA med 0,8 ml Buffer QF forvarmet ved 50 ° C.
- Precipitere DNA ved å tilsette 0,56 ml (0,7 volum elueringsbuffer) av romtemperaturisopropanol til det eluerte DNA.
- Bland ved inversjon (10X) og sentrifuger umiddelbart i 20 minutter ved 15.000 xg og 4 ° C. OmsorgFjern supernatanten helt uten å forstyrre den glasagtige, løst festede pellet.
- Vask den sentrifugerte DNA-pellet med 1 ml kald 70% etanol. Sentrifuger i 10 minutter ved 15.000 xg og 4 ° C.
- Fjern forsiktig supernatanten (vær også forsiktig med dette trinnet) uten å forstyrre pelleten. Lufttørke i 5-10 minutter og resuspendere DNA i 0,1 ml elueringsbuffer (EB). Oppløs DNA natten over ved romtemperatur. Unngå pipettering, noe som kan forskyve DNA.
- Kvantifiser prøvene ved hjelp av en høysensiv DNA-kvantifiseringsanalyse (per produsentens instruksjoner).
- Total DNA-ekstraksjon ved bruk av kommersielle DNA-ekstraksjonskolonner
- Perlebasert fraksjonering av metylert og umetylert DNA
MERK: En nylig publisert demonstrasjon av bruken av et kommersielt tilgjengelig sett 28 som utnytter en nedtrekksmetode som benytter et CpG-spesifikt metylbindende domeneprotein fusjonert til det humane IgG Fc-fragmentet (MBD2-Fc-protein) til fraksjonSpiste planteorganellgener (unmethylert) fra atomgenomet (høyt metylert) innhold 23 . Fraksjoneringseffektivitet i hveteprøver ble ikke tidligere testet ved bruk av dette kommersielle MF-settet 28 .- Ting å gjøre før du starter
- Tilbered frisk 80% etanol (minst 800 μl per reaksjon). Sett 5x bind / vaskebuffer for å tine på is og lag 5 ml 1x buffer per prøve (fortynnet 5X buffer med sterilt nukleasefritt vann og hold på is under protokollen).
- Klargjør MBD2-Fc proteinbundne magnetiske perler
- Forbered det nødvendige antall perlesett. Skal reaksjonene å bruke mellom 1 og 2 μg totalt inngangsd DNA, som krever 160 - 320 μl perler. Legg merke til at reaksjonene som er oppført nedenfor, gjelder for 1 μg av totalt inngangsd DNA, slik at de krever 160 μl perler. Skala reaksjonene i henhold til behovene.
- Ved hjelp av brede boretips pipetter du forsiktig protein A Magnetic BEad slurry opp og ned for å skape en homogen suspensjon. Som et alternativ, roter forsiktig rørene av perler i 15 minutter ved 4 ° C.
MERK: Ikke vortex perlene. - Fortsett med instruksjonene i henhold til produsentens instruksjoner 28 .
- Capture methylated nuclear DNA
- For hver enkelt prøve, tilsett 1 μg inngangsd DNA til et rør som inneholder 160 μl MBD2-Fc-bundet magnetiske perler.
- Tilsett 5x bind / vaskebuffer etter behov gitt volumet av DNA-inngangsprøven for en endelig konsentrasjon på 1x (volum 5x bind / vaskebuffer for å legge til (μL) = volum av inngangsd DNA (μL) / 4). Pipett prøven opp og ned noen ganger for å blande ved hjelp av en brede pipettespiss.
- Roter rørene ved romtemperatur i 15 minutter. Prøv forsiktig pipettprøvene med en brettet pipettespiss, og prøv prøvene 2-3 ganger i løpet av inkubasjonen for å forhindre perlklumping.
MERK: pipetteringen og flickienNg er kritisk for å sikre effektiv pulldown av det metylerte DNA.
- Samle beriket, ikke-metylert organellært DNA
- Spreng kort røret som inneholder DNA- og MBD2-Fc-bundet magnetisk perleblanding. Plasser røret på et magnetstativ i minst 5 minutter for å samle perlene til siden av røret. Løsningen skal vises tydelig.
- Ved hjelp av brede boretråder, fjern nøye den rydde supernatanten uten å forstyrre perlene. Overfør supernatanten (inneholder ikke-metylert, organellarberiget DNA) til et rent, lavt bindende, 2 ml mikrocentrifugerør. Oppbevar denne prøven ved -20 eller -80 ° C, eller fortsett direkte til trinn 3.2.6 for rensing.
- Elute fanget nukleært DNA fra MBD2-Fc-bundne magnetiske perler
- Hvis den nukleare fraksjonen også er ønsket, følg produsentens instruksjoner 28 for å eluere det nukleære DNA fra de MBD2-Fc-bundet magnetiske perler; Rens som beskrevet i trinn 3.2.7.
- Perlebasert nukleinsyrerensing
- Pass på at rensekulene er ved romtemperatur og blandes grundig. Fortsett med protokollen i henhold til instruksjonene i MF-kassehåndboken 28 .
MERK: Prøven kan nå brukes til NGS-bibliotekskonstruksjon eller annen nedstrømsanalyse.
- Pass på at rensekulene er ved romtemperatur og blandes grundig. Fortsett med protokollen i henhold til instruksjonene i MF-kassehåndboken 28 .
4. Eksempelkvantifisering og kvalitetskontroll
- QPCR-analyse for å vurdere organellarberikning
MERK: De qPCR-reaksjons- og analyseparametrene som er oppført her, ble konstruert for bruk på en Roche LightCycler 480 og må kanskje justeres for forskjellige utstyr og reagenser. Hvis qPCR ikke er tilgjengelig, kan endpoint-PCR og visualisering på en agarosegel brukes som et kvalitativt mål for prøverenhet ved bruk av de samme primere og betingelser beskrevet her. Amplicon størrelser vil være ~ 150 bp for alle primersett. Se tabell 3 for primer-sekvensenCes og parringer.- QPCR reaksjonsoppsett
- For å sette opp en individuell 20 μl qPCR-reaksjon, pipetteres følgende nøye i en enkelt brønn i en 96-brønn qPCR-plate: 10 μL 2x SYBR Green I Master; 2 μL av 10 μM forover- og revers-primerblandingen (for en sluttkonsentrasjon på 0,5 μM); 2 μL mal (innenfor standardkurven); og 6 ul sterilt, nuklease-fri H2O For å redusere pipetteringstrinn feil, er det å foretrekke å lage et konsentrat-blandingen med alle reaksjonskomponentene bortsett fra malen. Legg til masterblandingen på qPCR-platen, og legg deretter til malen av interesse for hver brønn. Tre tekniske replikater for hver prøve skal utføres for å minimere effekten av pipetteringsfeil.
MERK: Til slutt sammenlignes forholdet mellom atom- og organellar-kvantifiseringssykluser mellom prøver, så små forskjeller i konsentrasjon er akseptable. DNA-konsentrasjonene bør imidlertid være omtrent innenfor området eaCh andre. - Tetning platen med en qPCR-tetningsfilm av høy kvalitet. Vortex prøvene forsiktig, pass på å unngå å opprette noen bobler. Snur ned platen kort i 2 minutter ved 4 ° C for å samle prøven og fjern eventuelle små bobler.
- Legg platen i maskinen. Kjør qPCR-programmet i henhold til retningslinjene nedenfor.
- For å sette opp en individuell 20 μl qPCR-reaksjon, pipetteres følgende nøye i en enkelt brønn i en 96-brønn qPCR-plate: 10 μL 2x SYBR Green I Master; 2 μL av 10 μM forover- og revers-primerblandingen (for en sluttkonsentrasjon på 0,5 μM); 2 μL mal (innenfor standardkurven); og 6 ul sterilt, nuklease-fri H2O For å redusere pipetteringstrinn feil, er det å foretrekke å lage et konsentrat-blandingen med alle reaksjonskomponentene bortsett fra malen. Legg til masterblandingen på qPCR-platen, og legg deretter til malen av interesse for hver brønn. Tre tekniske replikater for hver prøve skal utføres for å minimere effekten av pipetteringsfeil.
- QPCR-reaksjonsparametere
MERK: Disse er standardparametere, bortsett fra annealing-syklusen i forsterkningsfasen. Juster denne innstillingen for å imøtekomme bestemte primere hvis de som brukes, er forskjellige fra de primere som presenteres i denne protokollen.- Forinkuber ved 95 ° C i 5 minutter, med en rampe på 4,4 ° C / s.
- Utfør 45 forsterkningssykluser på (1) 95 ° C i 10 s, med en rampe på 4,4 ° C / s; (2) 60 ° C i 20 s, med en rampe på 2,2 ° C / s; Og (3) 72 ° C i 10 s, med en rampe på 4,4 ° C / s (data oppnådd under (3)).
- Bruk en optioNal smeltekurve syklus på 95 ° C i 5 s, med en rampe på 4,4 ° C / s; 65 ° C i 1 min, med en rampe på 2,2 ° C / s; Og 97 ° C, med kontinuerlig oppkjøpsmodus.
- Bruk en kjølesyklus på 40 ° C i 30 s, med en rampe på 1,5 ° C / s.
- Analyseparametere
- Velg SYBR-malen. Kontroller programparametrene i Eksperiment-knappen. Når platen er lastet, kan analysen startes, og innstillingene kan justeres mens analysen kjører.
- Tilordne prøver ved hjelp av prøveditoren. Velg Abs Quant som arbeidsflyt og betegne prøvene som ukjente, standarder eller negative kontroller. Utpeker replikater og fyll inn prøvenavnene til den første av hver replikat. Legg til konsentrasjoner og enheter til standardene.
- Sett opp delinnstillinger for analyse; Disse tilordnes i delsetteditoren.
- For analyse, velg Abs Quant / 2nd Derivative Max fra listen "create new analysis".Importer den eksternt lagrede standardkurven (hvis aktuelt) og trykk deretter på beregne; Rapporten inneholder informasjonen som er valgt.
- For å utføre nøyaktig absolutt kvantifisering for bestemmelse av kopiantal eller konsentrasjon, bruk en standardkurve som er representativ for prøven som testes ( f.eks. Organellært DNA isolert fra metodene ovenfor). Siden mengden av mitokondrielt DNA som kreves for å lage en standardkurve er for høy til å bli oppnådd med en rimelig mengde vev, må du ikke bruke kopieringsberegninger som leveres av programvaren, men i stedet undersøke krysspunktspunkt (Cp) verdier for å bestemme den relative anrikningen Av organellar sammenlignet med kjernefysisk DNA i prøvene. Sammenligne disse relative mengder med de av totalt genomisk DNA (se representative resultater ). Prøveprimereffektivitet på fem 1:10 fortynninger av totalt genomisk DNA fra fullt lette, to ukers gamle hveteplanter (representativ effektivitet rapportert iE legende på figur 2 ).
- QPCR reaksjonsoppsett
- Pulsed-field gelelektroforese (PFGE)
MERK: Denne protokollen er basert på produsentens retningslinjer for å utføre PFGE for å løse DNA med høy molekylvekt. Se materialebordet.- Klargjør gelen og prøvene
- Følg retningslinjene for gel- og prøvepreparasjon og tilpass dem til det tilgjengelige systemet.
- Kjør parametrene
- Følg retningslinjene for oppsett av elektroforeseanlegget og bruk følgende parametere: Starttid for brytertid på 2 s, sluttbrytertid på 13 s, kjøretid på 15 timer og 16 min, V / cm på 6 og inkludert vinkel på 120 ° .
- Flekk og bilde gelen
- Farg gelet med et valgfritt fargestoff ( f.eks. Etidiumbromid eller et egnet alternativ) og bilde med et egnet geldokumentasjonssystem.
- Klargjør gelen og prøvene
- Bruk 1 ng DNA som inngang for DNA Library Prep Kit, etter produsentens instruksjoner.
- Strekkode og samle prøver for sekvensering i en enkelt runde. Utfør sekvensering i henhold til produsentens retningslinjer.
MERK: Pooling og sekvenseringsparametere kan endres avhengig av arten av interesse, ønsket dekningsnivå og plattformen som brukes til å sekvensere bibliotekene. For eksempel har en HiSeq-bane vesentlig mer utgang enn en MiSeq-bane, så mange flere prøver kan multiplexeres. Sequence en mindre delmengde av prøver for å avgjøre om dekningsnivåene av organellargenene er tilstrekkelige for nedstrømsanalyse.- Undersøk lesekvaliteten ved hjelp av FastQC 31 for å bestemme omfanget av trimning og filtrering som kreves for dataene.
- Trim og filtrer den råen leser ved å bruke Trimmomatic 32 eller et annet tilsvarende program. Bruk følgende innstillinger: ILLUMINACLIP 2:30:10 (for å fjerne adaptere), LEADING 3, TRAILING 3, SLIDINGWINDOW 4:10 og MINLEN 100.
- Merk at kvalitets filtrert og adapter-trimmet sammenkoblet ende (PE) leser til kinesisk Spring mitokondriell (NCBI Referanse Sekvens NC_007579.1 33), kloroplast (NCBI Referanse Sekvens NC_002762.1 34), og kjernefysiske 35 referanse genomer ved hjelp av Bowtie2 36, Med følgende innstillinger: -I 0 -X 800 - følsom.
- Konverter samlineringsfilene til bam-format (samtools) og sorter bam-filene. Bruk bam-filene til å beregne genoom-bred dekning og per-basis dekning med bedtools. Visualiser resultatene med R-plot-funksjonen.
- Ting å gjøre før du starter
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Protokollene som presenteres i dette manuskriptet beskriver to forskjellige metoder for å berike for organellært DNA fra plantevev. Forholdene som presenteres her reflekterer optimalisering for hvetevev. En sammenligning av viktige trinn i protokollene, nødvendig vevinngang og DNA-utgang er beskrevet i figur 1 . Trinnene i DC-protokollen vi testet følger lignende forhold til de som tidligere er beskrevet ( Figur 1A ). Høstet vev må behandles fersk og utsatt for differensial sentrifugering og / eller gradienter for å isolere intakte organeller. Nukleært DNA elimineres før organeller lyseres, og til slutt ekstraheres DNA'et og brukes til nedstrømsapplikasjoner. I motsetning, i MF-protokollen, kan plantevev høstes og lagres før bruk, og intakte organeller er ikke påkrevd. I stedet er kjernefysisk og organellært DNA fraksjonert fra totalt gDNA basert på DNA-metyleringsstatusen. Begge protokollene gir omtrent like store mengder organellært DNA ( figur 1C ). I forhold til total organellar DNA-utgang i forhold til vevsinngang, er MF-protokollen fordelaktig når vev er begrenset, ettersom en liten prøve fra en enkelt plante kan bli brukt, og planten kan få lov til å vokse for videre analyse. Vanligvis, i DC-protokoller, er alle luftvev fra mange frøplanter krevd, og disse plantene kasseres. Likevel kan DC-metoden optimaliseres for å spesifikt berike for en organeltype over den andre, noe som ikke er mulig med MF-tilnærmingen. Det er verdt å nevne at total tid for hver protokoll er omtrent likeverdig, selv om det er mindre praktisk tid i MF-tilnærmingen.
Begge metodene beriker for organellært DNA, om enn med forskjellige forhold av myokondrier og plastid-sekvenser:
"> Svært lave mengder renset organellært DNA er oppnådd ved hjelp av begge metoder (i størrelsesorden 50-100 ng, Figur 1C ). For å vurdere nivåene av organellært genom-anrikning og forurensning av atomgenomet i DNA isolert fra både DC og MF Metoder, ble en qPCR-analyse anvendt. I denne analysen ble de relative mengder av tre amplikoner ( dvs. nukleinspesifikke, ACTIN , mitokondrialspesifikke, NAD3 ; Kloroplast-spesifikk, PSBB ) ble vurdert i totalt genomisk DNA, og organellar DNA-fraksjonen ble oppnådd fra begge metoder ( figur 2 ). Kvantifikasjonssyklus (C q ) -verdier ble undersøkt for hver prøve ( figur 2A ), og fordi C q er definert som PCR-syklusen ved hvilken fluorescensen fra målforsterkningen øker over bakgrunnsfluorescensnivået, har C q og mål overflod en Omvendt forhold. IDC-prøven, C q av NAD3 og PSBB er henholdsvis ~ 17 og ~ 15 sykluser tidligere enn ACTIN (som har en C q på ~ 36) (se figur 2B for C q- verdier og anrikningsnivåer). Dette tilsvarer teoretiske 167.181 og 47.790 ganger anrikninger for henholdsvis NAD3 og PSBB , I forhold til ACTIN i DC-prøven ( figur 2B , se legenden om figur 2 for beregningen). I den totale genomiske DNA-prøven er de brette anrikninger for NAD3 og PSBB i forhold til ACTIN kun henholdsvis 158 og 10701. Det er ikke overraskende å finne en høyere overflod av organellaramplikonene i forhold til nukleær amplikon i totalt genomisk DNA, gitt at organellargenene eksisterer i større kopi-tall pr. Celle enn nukleærgenomet 37 og at antall organeller peCelle kan variere avhengig av vevstype eller utviklingsstadiet 38 , 39 . Samlet indikerer dataene at DC-metoden fortrinnsvis beriker for mitokondrier, som man kan forvente, da sentrifugeringshastigheter er optimalisert for selektivt å isolere mitokondrier og redusere atom- og kloroplast "forurensning."Den ummetylerte fraksjonen av MF totalt gDNA viser også betydelig anrikning av begge organellære amplikoner og forventes å beholde de relative relative mengder av disse målene. Foldberikningene for NAD3 og PSBB i forhold til ACTIN i den ummetylerte fraksjon er henholdsvis 20.551 og 1.703.253 ( Figur 2A og 2B ). I den metylerte fraksjon er de brette anrikninger for NAD3 og PSBB i forhold til ACTIN henholdsvis 31 og 823, indiCating at MBD2-Fc protein er svært effektiv ved utløpet av metylert nukleært DNA. Ettersom kloroplast amplikon har en høyere overflod enn mitokondriell amplikon i totalt genomisk DNA (~ 6 C q tidligere), metylert fraksjon (~ 5 C q tidligere) og umetylert fraksjon (~ 6 C q tidligere), antyder dette at Innfødt overflod av disse amplikonene blir ikke vesentlig endret ved MDB2-nedtrekking. Vi fokuserer her på den ikke-metylerte (organellære) fraksjonen på grunn av interessen for å sekvensere disse genomene spesifikt. Imidlertid, dersom kjernekomometet er hovedinteressen, vil MF og etterfølgende sekvensering av den metylerte fraksjon gi en mye høyere kjernemetom dekning enn total genomisk DNA-sekvensering, på grunn av reduksjonen i organellær DNA "forurensning."
Det er verdt å merke seg at hvis qPCR ikke er tilgjengelig, gir sluttpunkts-PCR (ved hjelp av de samme primere som for qPCR) kvalitaTiv vurdering av organellar renhet. I dette tilfelle vil rene organellære DNA-prøver vise amplifikasjon for mitokondrielle og plastid amplikonene, men ingen detekterbar amplifisering av nukleær amplikon på agarosegelen, mens totalt genomisk DNA viser amplifikasjon for alle tre primersettene, som vist i tidligere studier 11 , 12 .
Organellar DNA isolert fra begge metoder er egnet for NGS:
Trimmet og rengjort PE sekvensering leser (se trinn 4.3) ble kartlagt til tidligere publiserte hveteorganellar referanse genomer, og mengden av leser som ble brukt for kartlegging av hver prøve varierte fra ~ 800.000 til 1.100.000 leser ( Figur 3I ). Resultater fra kartlegging de novo Illumina-sekvensering leser til tilgjengelige hvetekloroplast og mitokondriergener er konsistente med qPCR resUlts, med DC-metoden som gir DNA som er mer beriket i mitokondrielt DNA ( Figur 3A og 3B , ~ 80% og 10% av leser kartet til henholdsvis mitokondrie (mt) og kloroplast (cp) genomene) og MF-metoden Gir DNA som sannsynligvis reflekterer den opprinnelige overflaten av de to organellære genomene ( figur 3A og 3B , ~ 20% og ~ 80% av leser kartet til henholdsvis mt og cp-genomene). I begge metodene overgår de teoretiske dekningene (se legenden til figur 3 for beregning) av begge hveteorganellargener over 100X dekning (og varierer opp til ~ 2000x dekning for kloroplastgenomet i den ikke-metylerte fraksjonen fra MF-metoden), selv Når 12 biblioteker er multiplexert ( figur 3C og 3D ; de 6 bibliotekene som er inkludert i denne analysen ble samlet sammen med ytterligere 6 biblioteker for en egen analyse, for totalt 12 bibliotekerSamlet i en enkelt sekvenseringsbane). En mer detaljert oppfatning av dekningen ble oppnådd ved å undersøke brøkdelen av genomet dekket på bestemte dybder, samt ved dokkingsnivåer per base ( Figur 3E -3I ). For MF-metoden var gjennomsnittlig per basis dekning ~ 300 - 450X for mt-genomet og 4.000 - 5.000X for cp-genomet. For DC-metoden var gjennomsnittlig per basis dekning ~ 900 - 1.300 og ~ 500 - 700X for henholdsvis mt og cp genomene. Det var imidlertid en liten brøkdel av både mt- og cp-genomene som hadde ekstremt lav eller høy dekning, og dette ble sett i organellært DNA avledet fra begge metoder ( figur 3I ). Regioner med høyere enn gjennomsnittlig dekning svarer sannsynligvis til regioner med homologi mellom organellargenene, og regioner med lav dekning kan indikere SNPs eller andre små varianter mellom de sorterte sekvensene og de publiserte referansene. Til støtte for denne oppfatningen, disse piggerAv høy dekning var mest uttalt for mt-DNA-avledet fra MF-metoden ( figur 3E og 3I ), sannsynligvis på grunn av den høye dekning av cp-genomet i denne metoden. Uforklarlig er dekningen av cp-genomet mer ujevn i MF-metoden enn DC-metoden ( Figur 3G og 3H ), som kan skyldes små forstyrrelser i MBD2-Fc-pulldown langs cp-DNA. Ytterligere eksperimenter vil bli pålagt for å avgjøre hvorfor dette er tilfelle. Uansett hadde mt- og cp-genomene relativt jevn dekning med begge metoder og ingen store områder med manglende dekning, hvilket kan påvises ved undersøkelse av fraksjonen av genomene sekvensert ved en gitt dybde ( figur 3E -3H ). I tillegg betraktes dekningsnivåene for begge genomene tilstrekkelig for nedstrømsanalyse, slik som variantanalyse. Hvis det anses nødvendig for analyse av sjeldne varianter, reduserer numbeR av samleprøver vil oppnå større dekning. Alternativt kan et langt større antall prøver bli samlet på en HiSeq-bane mens de oppnår enda større sekvensdybde, om enn ved et offer i sekvenslengde, idet HiSeq-biblioteker for tiden er begrenset til PE150-lengden i motsetning til PE300 MiSeq-biblioteker.
For å undersøke nivåene av forurensning av nukleærgener ved hjelp av en kartleggingsmetode, ble PE-lese kartleggingskategorier undersøkt. PE-leser kan kartlegges til et referansegenom i en rekke konfigurasjoner. Når man leser 1 og 2, retter man seg til referansen i hodet mot hodet, med en viss "forventet" avstand mellom de to vennene (basert på den gjennomsnittlige innskriftsstørrelsen på biblioteket og vanligvis angitt som en inngangsparameter i kartleggingsprogramvaren ), Disse PE leser sies å kart "concordantly." I motsetning er "uhensiktsmessig" kartlegging situasjonen der vennekartene har en mindre eller større enn forventet disTanse til referansegenomet eller kartet i alternative konfigurasjoner (fra hode til hale eller hale til hale). Hvis bare en kompis justerer seg til referansegenomet, sies det at den PE leses ikke verken sammenhengende eller uforskammet til referansegenomet. I alle tre lesekartkategorier kan PE-lesene justere seg til referansegenetet en eller flere ganger.
For både DC- og MF-isolert organellært DNA var lesekartlegging til mitokondriegenomet overveiende i den tilstrekkelige enverdige kategorien ( Figur 4A ), mens leser kartlagt til kloroplastgenomet i relativt like store mengder av enstemmig én gang og i samsvar med mer enn En gang ( figur 4B ), sannsynligvis på grunn av de store inverterte gjentagelser som er tilstede i kloroplastgenomet og også til de ekstremt høye dekningsnivåene. Færre PE leser imidlertid kortlagt til kjernefysisk genom og i stor grad kartlegges mer enn en gang i aVerken konkordant eller uheldig mote ( dvs. kun en kompis kan kartlegge). Disse er mest sannsynlig å kartlegge "off-target" til sekvenser i det nukleære genomet, som er homologe med organellargenene eller misassemblede regioner. Bare en mindre mengde av lyder (<5%) tilordnet den nukleære genom concordantly, noe som indikerer lave nivåer av kjerne genomet forurensning i organellar DNA isolert fra likestrøms eller MF-metoden (figur 4C), som gjenspeiles også qPCR resultater (figur 2A ). Nukleær fraksjon etter MBD2-Fc nedtrekking fra kinesisk vår ikke-etiolert vev ble også sekvensert for å bestemme hvor effektiv utløpet er ved fjerning av ummetylert DNA. Mindre enn 1% av leser i kjernefysisk fraksjon-avledet bibliotek kartlagt til organellar referanse genomene, mens ~ 45% av alle leser kartlagt til nukleært genom ( Figur 4 ). Men de fleste leser kartlagt på en uheldig måte, wSom sannsynligvis reflekterer de høye nivåene av misassembly og fragmentering i hvete-kjernevirksomhetsreferansen. Uansett, antyder resultatene at MBD2-Fc pulldown er svært effektiv ved fjerning av ummetylert organellært DNA fra metylert nukleært DNA. Det er verdt å merke seg at fordi det organellarberikte DNA som følger av disse metodene inneholder en blanding av mitokondrier og kloroplast-sekvenser, og fordi sekvenslikheter som følger av gammelt genoverføring mellom disse organeller forblir i deres genomene, vil riktig tildeling av leses til den spesifikke Genomene må løses bioinformatisk.
Etiolering av bladvæv endrer ikke anstendig organelle overflod:
Tradisjonelt foretrekkes etiolert vev for plante mitokondriell DNA-isolasjon for å redusere nivåene av fenol og stivelse, noe som kan forstyrre ekstraksjonenN eller nedstrøms applikasjoner 13 . For å bestemme om organellargenomberikningsnivåer kunne bli endret eller forbedret ved vekstbetingelser, ble både etiolerte og ikke-etiolerte vev utsatt for MF-protokollen og sekvensering. Interessant forandret etiolering ikke signifikant prosentandelen av lesene som mappet til organellarreferansegenomene ( figur 3A og 3B ) eller per-basisdekning ( Figur 3I ) sammenlignet med ikke-etiolerte betingelser. Vi isolerte også organellar DNA ved hjelp av differensial sentrifugering, med både etiolerte og ikke-etiolerte vev, og liten forskjell i anrikning ble funnet mellom de forskjellige vevene ved hjelp av qPCR (data ikke vist). Dette antyder at mer fysiologisk relevante ikke-etiolerte vev kan brukes til organellære sekvenseringsstudier uten merkbar forandring av anrikning.
Kvalitetskontroll foreslår detMF DNA er mest egnet for langlest sekvensering:
Siden langlest sekvensering blir mer tilgjengelig for forskere, blir isoleringen av høymolekylært DNA stadig viktigere. For å vurdere organellært DNA isolert med enten metode for intakt og kvalitet ble PFGE anvendt. Total genomisk DNA migrerer vanligvis som et diffust smear i PFGE, og molekylvekten bestemmes av protokollen og hvordan DNA ble lagret og håndtert etter ekstraksjon. Total genomisk DNA isolert med genomiske tips bør overstige 50 kb, som ble verifisert ved hjelp av PFGE ( figur 5 , felt 2). Det totale genomiske DNA fra de genomiske tipsene brukes som inngangen til Microbiome Enrichment Kit for å fraksjonere kjernen fra organellar DNA. Nukleærfraksjonen som oppnås etter fraksjonering, reduseres i størrelse, men gjenstår sentrert rundt 50 kb ( figur 5 , felt 4). Dette er ikke suGitt at den forholdsvis grovere håndtering av nukleær fraksjon som eluering fra MBD2-Fc-bundet perler krever varme- og proteinase K-fordøyelse. På grunn av den begrensede massen ble organellarfraksjonen ikke kjørt på PFGE, men etterfølgende analyse med TapeStation indikerte DNA> 50 kb (data ikke vist). Det organellære DNA oppnådd ved differensial sentrifugering har en gjennomsnittlig masse på ~ 20 kb, sannsynligvis forårsaket av den utvidede organellære isolasjonsprotokollen og den påfølgende kolonnebaserte DNA-ekstraksjon og konsentrasjon. Gradientbasert organellarisolasjon og alternative DNA-ekstraksjonsmetoder kan opprettholde større DNA-fragmentstørrelser. Uansett kan DNA av størrelsen oppnådd i denne protokollen brukes til å generere 10- eller 15-kb-sekvensering, leser om det tas hensyn under bibliotekets fremstilling.
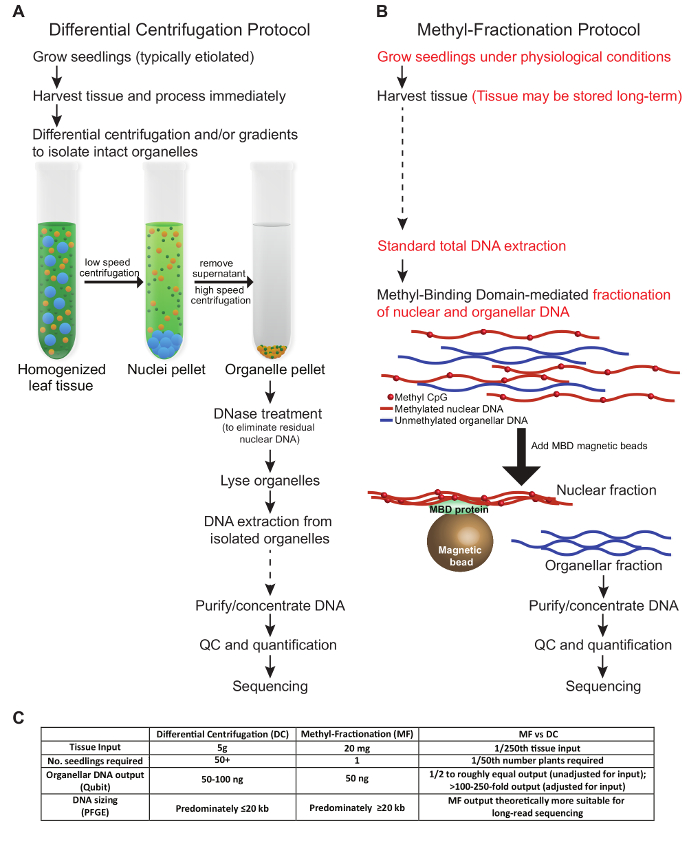
Figur 1: En sammenligningsvisning av to MethoDs til berikelse for planteorganell DNA. En tradisjonell DC-protokoll ( A ) er i motsetning til MF-protokollen ( B ). Det anbefales å unngå frysing og tining av prøvene. Trinnene der prøvene kan lagres på lang sikt, er imidlertid angitt med stiplede piler ( A og B ). Viktige forskjeller mellom protokollene er uthevet i rødt ( B ). ( C ) Tabellen sammenligner metodene i form av vevsinngang, antall planter som kreves, DNA-utgang og resulterende DNA-størrelse. Vennligst klikk her for å se en større versjon av denne figuren.

Figur 2: Vurdering av kjernefysisk DNA-kontaminering i organellær DNA-isolert ved bruk av to metoder. (
( B ) Tabellen viser C q- verdiene, som er vist på grafen i ( A ), og brettet anrikning av organellar amplikonene i forhold til ACTIN . * Brett anrikning = 2 (Cq ACTIN - Cq Target) . Formelen antar en perfekt effektivitet på 2 for hver primer sett, siden den mindre deviatIon av hver primer sett fra 2 er ubetydelig og vil få liten effekt på beregningen og den generelle trenden ( ACTIN = 1,961, NAD3 = 1,95 og PSBB = 1,989). Primereffektivitetene ble evaluert ved å lage en standardkurve med en serie på fem 1:10 fortynninger av totalt genomisk DNA. Vennligst klikk her for å se en større versjon av denne figuren.

Figur 3: Les kartlegging og teoretisk dekning av kloroplast og mitokondrielle genomer. Prosentandel av leser kartlagt til mitokondrie ( A ) eller kloroplast ( B ) Kinesisk vår referanse genomene. Tilsvarende teoretisk dekning av den kinesiske vår mitokondriale ( C ) eller kloroplast ( D ) referansegenoMes, forutsatt at genomstørrelser på henholdsvis 450 og 135 kb beregnes ved å bruke de totale lese-tallene og prosentandelen av leser kartlegging til de forskjellige genomene. Genom-bred fordeling av dekning for organellært DNA fra MF-metoden ( E og G ) eller DC-metoden ( F og H ). Dataene i panelene E - H er hentet fra den kinesiske vårets etiolerte prøve, men alle andre prøver viste en lignende trend. ( I ) Gjennomsnittlig, laveste og høyeste per basis dekning for alle prøver i panelene A - D. Prøvemerker inkludert "E" betegner etiolerte prøver, og "NE" betegner ikke-etiolerte prøver. DC indikerer DNA isolert med differensials sentrifugeringsmetoden og Unmethylated indikerer DNA som er i den ummetylerte fraksjon etter nedtrekk med MBD2-Fc (MF-protokoll). Prøver merket "Chris" betegner hvete Triticum aestivum'Chris'. CS betegner prøver av hvete Triticum aestivum 'Chinese Spring. Merk: På grunn av sekvenshomologi mellom kloroplast, mitokondrier og nukleære genomer som skyldes gammel genoverføring mellom organellargenene samt mellom organellar- og atomgenomene, kan en liten prosentandel av råleser kartlegges til flere genomer. I tillegg leser det som ikke kartlegger enten organellar referansegenomet ikke er representert i denne figuren. Dermed er prosentandelen som vises her ( A og B ) ikke totalt 100%. Vennligst klikk her for å se en større versjon av denne figuren.

Figur 4: PE Les-kartlegging til hvetekjernen. Andel av kategorier av PE Les kartleggingstyper til mitokondrielle (A) , kloroplast (B) , eller kjernevirksomhet (C) Chinese Spring referanse genomene. - E betegner etiolerte prøver og - NE betegner ikke-etiolerte prøver. DC indikerer DNA isolert med differensial-sentrifugeringsmetoden. Umetylert indikerer DNA som er i den ummetylerte fraksjonen etter pulldown med MBD2-Fc i MF-protokollen, og metylert betegner nukleær fraksjon etter MBD2-Fc pulldown. Prøver merket "Chris" betegner hvete Triticum aestivum 'Chris.' CS betegner prøver av hvete Triticum aestivum 'Chinese Spring.' Unmapped leser vises ikke. Vennligst klikk her for å se en større versjon av denne figuren.
Oad / 55528 / 55528fig5.jpg "/>
Figur 5: Undersøkelse av DNA-kvalitet ved bruk av PFGE. Hedesomallisk genomisk DNA (bane 2), hveteorganellært DNA oppnådd ved differensial sentrifugering (bane 3) og nukleær fraksjon etter MF med MBD2-Fc nedtrekksmetode (bane 4) ble utsatt for PFGE på en 1% agarosegel med en 1 kb utvidet stige brukt som markør (baner 1 og 5). Vennligst klikk her for å se en større versjon av denne figuren.
| Buffernavn | Oppskrift | Merknader | Metode |
| STE buffer | 400 mM sukrose, 50 mM Tris pH 7,8, 20 mM EDTA pH 8,0, 0,6% (vekt / volum) polyvinylpyrrolidon (PVP), 0,2% (vekt / volum) okseserumalbumin (BSA), 0.1% (vol / vol) p-merkaptoetanol (BME) | Bufferblanding som inneholder bare sukrose, Tris og EDTA kan gjøres opp til en måned i forveien og holdes ved 4 ° C. PVP, BSA og BME bør tilsettes fersk til en alikvot av den nødvendige mengden buffer rett før bruk. | Metode # 1 |
| ST buffer | 400 mM sukrose, 50 mM Tris pH 7,8, 0,6% (vekt / volum) polyvinylpyrrolidon (PVP), 0,1% (vekt / volum) bovint serumalbumin (BSA) | Bufferblanding som inneholder bare sukrose og Tris kan gjøres opp til en måned i forveien og holdes ved 4 ° C. Merk at ST-bufferen ikke inneholder EDTA eller BME, og inneholder en lavere konsentrasjon av BSA. | Metode # 1 |
| DNase lager | 2 mg / ml DNase i 0,15 M NaCl til en bestandskonsentrasjon på 2 mg / ml | Oppbevar 200 ul alikvoter ved -20 ° C. For å klargjøre DNase-arbeidsløsning (200 μl DNase-løsning per prøve) seTabell 1 nedenfor. Se hele protokollen nedenfor for fullstendig informasjon om DNase-fordøyelsen. DNase-arbeidsløsningen skal tilberedes fersk. For å stoppe DNase reaksjonen kreves en 400 mM EDTA pH 8,0 løsning (sluttkonsentrasjon som er nødvendig for å stoppe reaksjonen er 0,2 M EDTA, se full protokoll for detaljer). | Metode # 1 |
| DNase arbeidsløsning | 0,25 mg / ml DNase og 20 mM MgCl2 i ST Buffer | Forbered fersk, 200 ul per prøve. Konsentrasjoner som vises, er endelig reaksjonsvolum, så blande: 62,5 ul 2 mg / ml DNase (basert på endelig 500 pl reaksjonsvolum), 4 ul 1 M MgCl2 (basert på 200 ul DNase-løsning volum), og 133,5 ul av ST-buffer for Et sluttvolum på 200 ul. | Metode # 1 |
| Lysis Buffer | 20 mM EDTA pH 8,0; 10 mM Tris pH 7,9; 500 mM guanidin-HCI; 200 mM NaCl; 1% Triton X-100; 0,5 mg / ml lyseringsenzymer fraTrichoderma harzianum | Bland alle ingrediensene unntatt lysende enzymer og lagre ved romtemperatur. Lysende enzymer bør tilsettes fersk til en liten alikvot til umiddelbar bruk. | Metode nr. 2 |
Tabell 1: Oppskrifter av hjemmelagde buffere og arbeidslager.
| Konsentrasjonsark | |||||||
| PRØVNAMN | Tom enhet vekt (g) | Vekt av fylt enhet (g) | Fylt volum (ul, fylt minus tomme vekter) | Vekt etter 1. spinn (20 min *, g) | Volum Etter 1. Spin (ul, fyltMinus tomme vekter) | Vekt etter 2. spinn (X min *, g) | Volum Etter 2. Spin (ul, fylt minus tomme vekter) |
| Merk at det faktiske gjenvunnet volumet vil være noen ul mindre enn beregnet volum. | |||||||
Tabell 2: Konsentrasjonsark.
| Navn | Genomspesifikitet | Gene Sequence Source | Sekvens (5 '- 3') |
| Ta_ACTIN - F | Nuclear | Gramene Stillas IWGSC_CSS_1AS_scaff_3272162: 10,663-12,557 | CAGGTATCGCTGACCGTATGA |
| Ta_ACTIN - R | Nuclear | Samme som ovenfor | GAAGGTAGGGCTGAACAAGAAAC |
| Ta_NAD3 - F | mitokondrie | NCBI tiltredelse EU534409.1 | GGTGATGCCAGAAGTCGTTT |
| Ta_NAD3 - R | mitokondrie | Samme som ovenfor | CAGATCAATCTTGTTAGGAGGTACTG |
| Ta_PSBB - F | kloroplast | NCBI-tiltredelse KJ592713.1 | GCTACCTTTGCTTTGCTCTTCT |
| Ta_PSBB - R | kloroplast | Samme som ovenfor | GCTGCCTGTTTCCTTGTAGTT |
Tabell 3: Liste over qPCR Primers.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Til dags dato, de fleste organellar sekvensering studier senter på tradisjonelle DC metoder for å berike for bestemt DNA. Metoder for å isolere organeller fra forskjellige planter har blitt beskrevet, inkludert mose 40 ; Monokotter som hvete 15 og havre 11 ; Og dikoter som arabidopsis 11 , solsikke 17 og rapsfrø 14 . De fleste protokoller fokuserer på bladvev 13 , 14 , 15 , 16 , 17 , med noen blitt tilpasset for en rekke vevstyper, inkludert frø 11 . Isoleringen av organeller fra protoplaster har også blitt påvist 41 . Dette er imidlertid ikke mottagelig for alle systemer, og det er heller ikke mulig når det er begrenset vev av interesse. Mange av disse orgaNellære isolasjonsmetoder ble konstruert for å gjenvinne intakte organeller for spesifikke eksperimenter, slik som fysiologiske studier. Disse protokollene er besværlige og krever vanligvis bruk av tetthetgradienter, slik som sukrose eller Percoll-gradienter, som er svært effektive ved isolering av spesifikke organellære fraksjoner, men krever et stort vevsinngang ( dvs. over 5 g og oppover i kilo, avhengig av Vevetypen). Likevel kan DC-metoden optimaliseres for å berike for spesifikke cellulære fraksjoner, så som mitokondrier eller kloroplast, ved å endre rotasjonshastigheter og tetthetgradienter. I motsetning krever MF-tilnærmingen langt mindre utgangsmateriale (20 mg), men mitokondrielle og plastid-DNA vil være tilstede per deres relative overflod i vevet som brukes for DNA-ekstraksjon. Ikke desto mindre tilbyr MF-protokollen en alternativ tilnærming for isolering av blandet organellært DNA og er spesielt gunstig for å starte med små mengder vev.
T O Vurder prøve renhet etter organelle isolasjon, de fleste studier til dags dato bare bruke sluttpunkt PCR og gelelektroforese 11 , 12 . Dette gir en rettferdig kvalitativ måling av prøverenhet. Imidlertid kan lave nivåer av amplifikasjon ikke bli visualisert på en agarosegel. Få rapporter inneholder mer kvantitative tiltak for kvalitetskontroll, som qPCR 14 . For en kvantitativ vurdering av DNA-prøve-renhet isolert fra begge metodene, benyttet vi qPCR og sekvensering for å bestemme hvor mye kjernek DNA forblir i prøven, samt de relative proporsjonene av mitokondrielt versus kloroplast DNA. Begge metodene som er evaluert her, er effektive for å fjerne kjernefysisk DNA. Begge metodene gir en blanding av mitokondrielt og chloroplast-DNA, om enn i forskjellige proporsjoner.
Voksende planter i mørket (etiolasjon) rapporteres å bidra til å lette organellarisolasjon på grunn av reduksjon av fenolRef "> 13. Men i denne sammenligningen fant vi ikke en merkbar fordel å arbeide med etiolert vev over lette dyr. Selv om andelen spesialiserte kloroplaster sannsynligvis vil være høyere når de er lette, blir det totale plastidnummeret, som Reflektert i andelen av leser kartlegging til kloroplastgenomet, er uendret under forskjellige lysforhold. Derfor anbefales det å utføre genomisk sekvensering på nedstrømsfunksjonelle analyser, som for eksempel vurdering av heteroplasm i forskjellige vev eller under forskjellige stressorer eller for ekspresjonsanalyser. Planter dyrket under fysiologisk relevante forhold.
For applikasjon med kortlest sekvenseringsteknologi, gir begge teknikkene her sammenlignet med tilstrekkelig DNA-kvantitet og kvalitet. For å oppnå lange lesninger på> 20 kb for enkeltmolekylære sekvenseringsapplikasjoner er imidlertid en større mengde DNA av høyere kvalitet nødvendig. For eksempel, ideelt,> 1 μg ren orgaNellar hvete-DNA med en molekylvekt> 20 kb er nødvendig for in-house, low-inngang protokoller for 20-kb innskudd bibliotek preparater 42 . Nye brukerutviklede, lavinngangsprotokoller kan redusere DNA-krav ( dvs. til 50 ng eller enda mindre 20 ), men utfordringen er fortsatt å ha høyverdig DNA med høy molekylvekt som går inn i bibliotekets preparater. Det er viktig at et flertall av DNA er> 20 kb, da mindre fragmenter vil bli fortrinnsvis satt inn i SMRTbell og kaste av størrelsesfordelingen av biblioteket 43 . Vi prøvde en rekke hjemmelagde DNA-ekstraksjonsprotokoller og en rekke kommersielle protokoller for DNA-ekstraksjon (ikke vist). For hvetebladvev ble den beste balansen mellom DNA-kvantitet og kvalitet, spesielt lengde, oppnådd ved anvendelse av et kommersielt sett 27 , 29 . Avhengig av plantearter og vev av interesse, alternatiVe ekstraheringsprotokoller kan være like egnet eller mer fruktbare. Likevel konkluderer vi at den totale ekstraksjon av genomisk DNA med en molekylvekt på> 50 kb, etterfulgt av fraksjonering med MBD2-Fc nedtrekks-tilnærming 28 , er egnet til langlestende sekvensering fra begrenset utgangsmateriale. Fremtidig arbeid bør teste grensene for utgangsmaterialet som kreves etter fraksjonering for langt innspillingsbibliotekspreparasjon og etterfølgende langlestsekvensering. Kritisk kunne denne tilnærmingen gi en robust metode for å isolere DNA fra en undersamling av et enkelt blad som er egnet for langlestende sekvensering, uten helgenomdannelse. Vi forventer at denne tilnærmingen lett kan tilpasses til flere vevstyper og i stor grad gjelder for andre plantearter. Det vil være spesielt nyttig i situasjoner der vevsmengden er begrensende, slik som sekvensering ved individuelle generasjoner i en kryssordning eller i sjeldnere vevtyper.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Forfatterne erklærer at de ikke har konkurrerende interesser.
Nevn av handelsnavn eller kommersielle produkter i denne publikasjonen er utelukkende for å gi spesifikk informasjon og innebærer ikke anbefaling eller påtegning fra US Department of Agriculture. USDA er en likestillingsleverandør og arbeidsgiver.
Acknowledgments
Vi ønsker å anerkjenne finansiering fra USAs Department of Agriculture-Agricultural Research Service og fra National Science Foundation (IOS 1025881 og IOS 1361554). Vi takker R. Caspers for vedlikehold av drivhus og plantepleie. Vi takker også Universitetet i Minnesota Genomics Center, hvor Illumina biblioteket forberedelser og sekvensering ble utført. Vi er også takknemlige for kommentarene fra journalredaktørene og fire anonyme anmeldere som styrket vårt manuskript ytterligere. Vi takker også OECD for et fellesskap til SK for å integrere disse protokollene for samarbeidsprosjekter med kolleger i Japan.
Materials
| Name | Company | Catalog Number | Comments |
| 2-mercaptoethanol (beta-mercaptoethanol; BME) | Sigma Aldrich | M3148-100ml | |
| 2-propanol (Isopropyl alcohol/isopropanol), bioreagent | Sigma Aldrich | I9516 | |
| agarose, Bio-Rad Cetified Megabase agarose | Bio-Rad | 1613108 | |
| analytical balance | Mettler Toledo | AB54-S | |
| balance | Mettler Toledo | PB1502-S | |
| bovine serum albumin (BSA) | Sigma Aldrich | B4287-25G | |
| Ceramic grinding cylinders, 3/8in x 7/8in | SPEX SamplePrep | 2183 | |
| Cryogenic Blocks compatible with tissue homogenizer for holding 50 mL tubes | SPEX SamplePrep | 2664 | |
| DNaseI | Sigma | DN25 | |
| ethanol, absolute | Decon Laboratories | 2716 | |
| Ethylenediamine Tetraacetic Acid (EDTA), 0.5 M Solution, pH 8.0 | Fisher | BP2482-500 | |
| gel imaging system | |||
| gel stain | Such as GelRed or Ethidium Bromide | ||
| grinding pestle, wide tip for 2 mL conical tubes | |||
| Guanidine-HCl, 8 M solution | ThermoFisher | 24115 | |
| LightCycler 480 SYBR Green I Master | Roche | 4707516001 | |
| liquid nitrogen | |||
| Lysing enzymes from Trichoderma harzianum | Sigma | L1412 | |
| Magnesium Chloride | G Bioscience | 24115 | |
| magnetic rack | ThermoFisher | A13346 | |
| microcentrifuge tubes, LoBind 1.5 mL | Eppendorf | 22431021 | |
| microcentrifuge tubes, standard nuclease-free 1.5 mL | Eppendorf | ||
| microcentrifuge, refrigerated | Sorvall | Legend X1R | Or equivalent product, must be capable of reaching at least 18,000 x g with rotors for 50 mL tubes, Oak Ridge tubes, and 1.5 mL tubes |
| microcentrifuge, room temperature | Eppendorf | 5424 | Or equivalent product, must be capable of reaching at least 18,000 x g with rotor for 1.5 mL and 2 mL microcentrifuge tubes |
| Microcon DNA Fast Flow Centrifugal Filter Units | EMD Millipore | MRCFOR100 | |
| Miracloth, 1 square per sample cut to fit funnel | EMD Millipore | 475855 | |
| NEBNext Microbiome DNA Enrichment Kit | New England Biolabs | E2612L | |
| parafilm | Parafilm M | PM992 | |
| plastic pots and trays | |||
| polyvinylpyrrolidone (PVP) | Fisher | BP431-100 | |
| Proteinase K | Qiagen | 19131 | |
| Pulsed-Field Gel Electrophoresis rig (e.g. CHEF DR III) | Bio-Rad | 1703697 | |
| purification beads, Agencourt AMpureXP beads | Beckman Coulter | A63881 | |
| QIAamp DNA Mini Kit | Qiagen | 51304 | |
| Qiagen 20/g Genomic Tip DNA Extraction Kit | Qiagen | 10223 | |
| Qiagen Buffer EB (elution buffer) | Qiagen | 19086 | |
| Qiagen DNA Extraction Buffer Set | Qiagen | 19060 | |
| QiaRack | Qiagen | 19015 | |
| qPCR machine (e.g. Roche Light Cycler 480) | Roche | ||
| qPCR plate sealing film | Roche | 4729757001 | |
| qPCR plate, 96 well plate | Roche | 4729692001 | |
| Qubit assay tubes | Life Technologies | Q32856 | |
| Qubit Broad Spectrum assay kit | Life Technologies | Q32850 | |
| Qubit High Sensitivity assay kit | Life Technologies | Q32851 | |
| RNaseA | Qiagen | 19101 | |
| Serological pipettes (20 mL) and pipet-aid | Fisher | 13-678-11 | |
| Small funnels, 1 per sample | |||
| Sodium Chloride | Ambion | AM9759 | |
| Soft paintbrush, 2 per sample | |||
| SPEX SamplePrep 2010 Geno/Grinder or another type of tissue homogenizer | SPEX SamplePrep | Or another comparable tissue homogenizer. If you do not have access to a tissue homogenizer, then grinding in a pre-chilled mortar and pestle will suffice (see protocol for details). However, a homogenizer will give more consistent results and total homogenization time is reduced. | |
| Sucrose | Omnipure | 8550 | |
| TBE | |||
| thermomixer | |||
| Tris | Sigma | T2819-100ml | |
| Triton X-100 | Promega | H5142 | |
| tube rotater | |||
| tubes, 50 mL conical polypropylene | Corning | 352070 | |
| tubes, 50 mL high-speed polypropylene | ThermoScientific/Nalgene | 3119-0050 | e.g. Nalgene Oakridge tubes or equivalent |
| vermiculite | |||
| water bath | |||
| water, sterile and certified Nuclease-free | Fisher | 1481 | |
| water, sterile milliQ |
References
- Liberatore, K. L., Dukowic-Schulze, S., Miller, M. E., Chen, C., Kianian, S. F. The role of mitochondria in plant development and stress tolerance. Free Radic Biol Med. 100, 238-256 (2016).
- Samaniego Castruita, J. A., Zepeda Mendoza, M. L., Barnett, R., Wales, N., Gilbert, M. T. Odintifier--A computational method for identifying insertions of organellar origin from modern and ancient high-throughput sequencing data based on haplotype phasing. BMC Bioinformatics. 16 (232), 1-13 (2015).
- Zhang, T., Zhang, X., Hu, S., Yu, J. An efficient procedure for plant organellar genome assembly, based on whole genome data from the 454 GS FLX sequencing platform. Plant Methods. 7 (38), 1-8 (2011).
- Wambugu, P. W., Brozynska, M., Furtado, A., Waters, D. L., Henry, R. J. Relationships of wild and domesticated rices (Oryza AA genome species) based upon whole chloroplast genome sequences. Sci Rep. 5 (13957), 1-9 (2015).
- Iorizzo, M., et al. De novo assembly of the carrot mitochondrial genome using next generation sequencing of whole genomic DNA provides first evidence of DNA transfer into an angiosperm plastid genome. BMC Plant Biol. 12 (61), 1-17 (2012).
- Park, S., et al. Complete sequences of organelle genomes from the medicinal plant Rhazya stricta (Apocynaceae) and contrasting patterns of mitochondrial genome evolution across asterids. BMC Genomics. 15 (405), 1-18 (2014).
- Skippington, E., Barkman, T. J., Rice, D. W., Palmer, J. D. Miniaturized mitogenome of the parasitic plant Viscum scurruloideum is extremely divergent and dynamic and has lost all nad genes. Proc Natl Acad Sci U S A. 112 (27), E3515-E3524 (2015).
- Wicke, S., Schneeweiss, G. M. Chapter 1. Next Generation Sequencing in Plant Systematics. Hörandl, E., Appelhans, M. , Koeltz Scientific Books. (2015).
- Sloan, D. B. One ring to rule them all? Genome sequencing provides new insights into the 'master circle' model of plant mitochondrial DNA structure. New Phytol. 200 (4), 978-985 (2013).
- Woloszynska, M. Heteroplasmy and stoichiometric complexity of plant mitochondrial genomes--though this be madness, yet there's method in't. J Exp Bot. 61 (3), 657-671 (2010).
- Ahmed, Z., Fu, Y. B. An improved method with a wider applicability to isolate plant mitochondria for mtDNA extraction. Plant Methods. 11 (56), 1-11 (2015).
- Ejaz, M., et al. Comparison of small scale methods for the rapid and efficient extraction of mitochondrial DNA from wheat crop suitable for down-stream processes. Genet Mol Res. 13 (4), 10320-10331 (2014).
- Eubel, H., Heazlewood, J. L., Millar, A. H. Isolation and subfractionation of plant mitochondria for proteomic analysis. Methods Mol Biol. 355, 49-62 (2007).
- Hao, W., Fan, S., Hua, W., Wang, H. Effective extraction and assembly methods for simultaneously obtaining plastid and mitochondrial genomes. PLoS One. 9 (9), e108291 (2014).
- Pomeroy, M. K. Studies on the respiratory properties of mitochondria isolated from developing winter wheat seedlings. Plant Physiol. 53 (4), 653-657 (1974).
- Taylor, N. L., Stroher, E., Millar, A. H. Arabidopsis organelle isolation and characterization. Methods Mol Biol. 1062, 551-572 (2014).
- Triboush, S. O., Danilenko, N. G., Davydenko, O. G. A method for isolation of chloroplast DNA and mitochondrial DNA from Sunflower. Plant Mol Biol Rep. 16 (2), 183-189 (1998).
- Pinard, R., et al. Assessment of whole genome amplification-induced bias through high-throughput, massively parallel whole genome sequencing. BMC Genomics. 7 (216), 1-21 (2006).
- Lamble, S., et al. Improved workflows for high throughput library preparation using the transposome-based Nextera system. BMC Biotechnol. 13 (104), 1-10 (2013).
- Raley, C., et al. Preparation of next-generation DNA sequencing libraries from ultra-low amounts of input DNA: Application to single-molecule, real-time (SMRT) sequencing on the Pacific Biosciences RS II. bioRxiv. , (2014).
- Tsai, Y. C., et al. Resolving the Complexity of Human Skin Metagenomes Using Single-Molecule Sequencing. MBio. 7 (1), e01948 (2016).
- Feehery, G. R., et al. A method for selectively enriching microbial DNA from contaminating vertebrate host DNA. PLoS One. 8 (10), e76096 (2013).
- Yigit, E., Hernandez, D. I., Trujillo, J. T., Dimalanta, E., Bailey, C. D. Genome and metagenome sequencing: Using the human methyl-binding domain to partition genomic DNA derived from plant tissues. Appl Plant Sci. 2 (11), 1-6 (2014).
- Noyszewski, A. K., et al. Accelerated evolution of the mitochondrial genome in an alloplasmic line of durum wheat. BMC Genomics. 15 (67), 1-16 (2014).
- Qiagen. QIAamp DNA Mini and Blood Mini Handbook. , 5th ed, Available from: https://www.qiagen.com/ch/resources/ (2016).
- E.M. Corporation. User Guide: Microcon Centrifugal Filter Devices. , Available from: http://www.emdmillipore.com/US/en/product/Microcon-DNA-Fast-Flow-Centrifugal-Filter-Unit-with-Ultracel-membrane,MM_NF-MRCF0R100 (2013).
- Qiagen. User developed protocol: Isolation of genomic DNA from plants and filamentous fungi using the QIAGEN Genomic-tip - (EN). , Available from: https://www.qiagen.com/ch/resources/ (2001).
- New England BioLabs, Inc.. NEBNext Microbiome DNA Enrichment Kit: Instruction Manual Version 4.0. , Available from: http://www.neb.com/~/media/Catalog/All-Products/371BCB5A557C462D95D1E45E15BBFEA3/Datacards or Manuals/E2612Manual.pdf (2015).
- Qiagen. QIAGEN Genomic DNA Handbook. , Available from: https://www.qiagen.com/ch/resources/ (2012).
- PacificBiosciences. Guidelines for Using the BIO-RAD® CHEF Mapper® XA Pulsed Field Electrophoresis System. , Available from: http://www.pacb.com/wp-content/uploads/Unsupported-Guidelines-Using-BIO-RAD-CHEFMapper-XA-Pulsed-Field-Electrophoresis.pdf (2016).
- Andrews, S. FastQC: A quality control tool for high throughput sequence data. , Available from: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (2016).
- Bolger, A. M., Lohse, M., Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 30 (15), 2114-2120 (2014).
- Ogihara, Y., et al. Structural dynamics of cereal mitochondrial genomes as revealed by complete nucleotide sequencing of the wheat mitochondrial genome. Nucleic Acids Res. 33 (19), 6235-6250 (2005).
- Ogihara, Y., et al. Structural features of a wheat plastome as revealed by complete sequencing of chloroplast DNA. Mol Genet Genomics. 266 (5), 740-746 (2002).
- International Wheat Genome Sequencing Consortium (IWGSC). A chromosome-based draft sequence of the hexaploid bread wheat (Triticum aestivum) genome. Science. 345 (6194), (2014).
- Langmead, B., Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat Methods. 9 (4), 357-359 (2012).
- Bendich, A. J. Why do chloroplasts and mitochondria contain so many copies of their genome? Bioessays. 6 (6), 279-282 (1987).
- Kumar, R. A., Oldenburg, D. J., Bendich, A. J. Changes in DNA damage, molecular integrity, and copy number for plastid DNA and mitochondrial DNA during maize development. J Exp Bot. 65 (22), 6425-6439 (2014).
- Ma, J., Li, X. Q. Organellar genome copy number variation and integrity during moderate maturation of roots and leaves of maize seedlings. Curr Genet. 61 (4), 591-600 (2015).
- Lang, E. G., et al. Simultaneous isolation of pure and intact chloroplasts and mitochondria from moss as the basis for sub-cellular proteomics. Plant Cell Rep. 30 (2), 205-215 (2011).
- Tobin, A. K. Subcellular fractionation of plant tissues. Isolation of chloroplasts and mitochondria from leaves. Methods Mol Biol. 59, 57-68 (1996).
- PacificBiosciences. Procedure & Checklist - 10 kb to 20 kb Template Preparation and Sequencing with Low (100 ng) Input DNA. , Available from: http://www.pacb.com/wp-content/uploads/Procedure-Checklist-10-20kb-Template-Preparation-and-Sequencing-with-Low-Input-DNA.pdf (2015).
- PacificBiosciences. Template Preparation and Sequencing Guide. , Available from: http://www.pacb.com/wp-content/uploads/2015/09/Guide-Pacific-Biosciences-Template-Preparation-and-Sequencing.pdf (2014).
