

Summary
Utilizing a preassembled Cas9 ribonucleoprotein complex (RNP) is a powerful method for precise, efficient genome editing. Here, we highlight its utility across a broad range of cells and organisms, including primary human cells and both classic and emerging model organisms.
Abstract
Site-specific eukaryotic genome editing with CRISPR (clustered regularly interspaced short palindromic repeats)-Cas (CRISPR-associated) systems has quickly become a commonplace amongst researchers pursuing a wide variety of biological questions. Users most often employ the Cas9 protein derived from Streptococcus pyogenes in a complex with an easily reprogrammed guide RNA (gRNA). These components are introduced into cells, and through a base pairing with a complementary region of the double-stranded DNA (dsDNA) genome, the enzyme cleaves both strands to generate a double-strand break (DSB). Subsequent repair leads to either random insertion or deletion events (indels) or the incorporation of experimenter-provided DNA at the site of the break.
The use of a purified single-guide RNA and Cas9 protein, preassembled to form an RNP and delivered directly to cells, is a potent approach for achieving highly efficient gene editing. RNP editing particularly enhances the rate of gene insertion, an outcome that is often challenging to achieve. Compared to the delivery via a plasmid, the shorter persistence of the Cas9 RNP within the cell leads to fewer off-target events.
Despite its advantages, many casual users of CRISPR gene editing are less familiar with this technique. To lower the barrier to entry, we outline detailed protocols for implementing the RNP strategy in a range of contexts, highlighting its distinct benefits and diverse applications. We cover editing in two types of primary human cells, T cells and hematopoietic stem/progenitor cells (HSPCs). We also show how Cas9 RNP editing enables the facile genetic manipulation of entire organisms, including the classic model roundworm Caenorhabditis elegans and the more recently introduced model crustacean, Parhyale hawaiensis.
Introduction
fThe CRISPR-Cas9 system allows scientists to alter targeted regions of any genome1. This quick and inexpensive technology has revolutionized basic research and promises to make a profound impact on the development of personalized disease therapies, precision agriculture, and beyond2. CRISPR editing is a democratizing tool and implementing the system in a new laboratory requires no particular expertise in genome engineering, just basic molecular biology skills. Researchers can now study previously intractable organisms with a few alternative means for genetic manipulation3,4. In the past five years alone, CRISPR genome editing has been used to engineer over 200 different vertebrates, invertebrate, plant, and microbial species.
Adapted from the CRISPR prokaryotic defense pathway, the core elements required for site-specific genome editing are the Cas9 protein, typically from S. pyogenes and codon-optimized with an added nuclear localization signal (NLS), and its specialized RNA guide5,6. Though not discussed here, other Cas9 orthologues or CRISPR endonucleases may also be used. The naturally occurring gRNA is composed of two separately transcribed pieces, the CRISPR RNA (crRNA) and the trans-activating crRNA (tracrRNA)7. These RNAs can be fused into a single transcript, known as the single-guide RNA (sgRNA)8. Most genome editors choose the streamlined sgRNA9, though the dual-guide is also used regularly10,11. Experimenters choose a 20-nucleotide (nt) genomic DNA target, ensuring that it lies next to a short licensing signature required for Cas9 recognition, called a protospacer adjacent motif (PAM), and design a gRNA that contains the complementary sequence12.
Once inside the cell, the RNP complex locates its genomic target, the gRNA base pairs with the complementary DNA strand, and then the enzyme cleaves both DNA strands to generate a double-strand break2. Cell repair machinery fixes the DSB by one of at least two routes: via the error-prone non-homologous end-joining (NHEJ) pathway or the homology-directed repair (HDR), which seamlessly incorporates DNA containing 'arms' of homology to either side of the break. The former repair pathway typically leads to indel formation and consequent gene disruption, while the latter allows experimenters to insert or change DNA sequences1.
The editing efficiency and accuracy depend on the means by which Cas9 and gRNA enter into the cell. These components may be delivered to cultured cells, embryos, or organisms in the form of nucleic acids or as a preassembled RNP complex13,14,15. Common nucleic acid-based delivery methods include the viral transduction, transfection, or electroporation of mRNA or plasmid DNA. Cas9 protein and guide RNA are then produced within the cell and they associate to form a complex.
The direct delivery of RNP requires the separate purification of the Cas9 protein and guide RNA. This can be done in-house, or the protein and sgRNA can be purchased from one of several commercial vendors. Once acquired, the Cas9 and gRNA are mixed to form the enzymatically-competent RNP complex and introduced to cells by direct injection into fertilized eggs/embryos, lipid-based transfection16, or electroporation. The first report of RNP editing involved injection into C. elegans gonads17. Microinjection is still the preferred means of introducing RNP into embryos and whole organisms, though effective electroporation has been demonstrated in mouse18,19 and rat20 embryos. We describe protocols for directly injecting RNP into C. elegans gonads and P. hawaiensis embryos and recommend a specialized type of electroporation to deliver RNP when editing primary human cells. This method, nucleofection, involves optimized electroporation programs and cell type-specific solutions and allows the RNP to enter both the cytoplasm and the nucleus21.
Genome editing with RNP offers several distinct advantages. Because the protein and RNA components are pre-assembled, and quality can be ensured prior to delivery, RNP editing avoids many pitfalls associated with the nucleic acid-based delivery. Namely, there is no risk of Cas9-encoding DNA integration into the host genome, mRNA is never exposed for degradation, and it circumvents problems with in vivo gRNA or protein expression, folding, and association22,23. Further, using RNP leads to lower toxicity and far fewer off-target events than the plasmid-based expression, a result of the RNP's shorter half-life inside the cell24,25,26,27.
Finally, RNP editing demonstrably leads to high editing rates in a variety of human cell lines, primary cells such as fibroblasts, embryonic stem cells (ESCs), induced pluripotent stem cells (iSPCs), HSPCs, and T cells16,24,25,26,27,28,29; in invertebrates including C. elegans, P. hawaiensis, and fruit flies3,17,30; in vertebrate species like zebrafish, mice, and rats31,32; in plant species including Arabidopsis, tobacco, lettuce, rice, grapevine, apple, maize, and wheat33,34,35,36; and in Chlamydomonas, Penicillium, and Candida species37,38,39. The frequency of indel formation can be higher when using RNP compared to the plasmid delivery, and HDR-mediated DNA insertion can be easier to achieve25,27,29.
The protocol described here uses the Cas9 RNP and is an effective, readily adaptable technique that is straightforward to apply to a wide variety of biological systems40,41, especially in cells that are otherwise difficult to work with and in organisms without well-established systems for precise genetic manipulation. We start by describing how to design, obtain, and assemble the Cas9 RNP before covering its use across different model cell types and organisms. Hematopoietic stem/progenitor cells (HSPCs) and T cells are edited using the same method, nucleofection, so they are covered together in steps 2 and 3 of this protocol. Editing procedures for C. elegans are described in steps 4 and 5, and P. hawaiensis editing is covered in steps 6 and 7. Finally, since the success of a gene-editing experiment in any organism may be assessed by genotype sequencing, substeps describing possible analysis methods for all the cells and organisms described in the protocol are outlined in step 8.
Protocol
1. RNP Assembly
- Design the experiment well in advance, acquiring all the RNA, DNA, and protein components ahead of time. As a first pass, try one of the positive controls listed in Table 1 and use the commercial reagents described in the Table of Materials to ensure a reliable experimental design and the integrity of the materials. For additional tips on planning a new genome-editing experiment, see papers on this topic12,42,43.
NOTE: Once assembled as described in the subsequent steps, RNPs prepared in advance may be stored at -80 °C.- After choosing which gene to target, use one of the free online tools to design an optimal gRNA44,45,46,47,48. Be sure to target an exon if hoping to generate a knockout.
Note: These tools will help to identify a target site with an adjacent S. pyogenes PAM sequence, high-quality score, and low off-target score. - Purify the S. pyogenes Cas9 protein through published methods8, or purchase it from a commercial vendor.
- Prepare a typical Cas9 buffer for the RNA dilution, RNP preparation, and protein storage, which contains 20 mM of HEPES pH 7.5, 150 mM of KCl, 10% glycerol, and 1 mM of TCEP. Always use nuclease-free water in buffers that will be used to resuspend or dilute RNA to prevent degradation.
- Produce the guide RNA (tracrRNA and crRNA or sgRNA) through an in vitro transcription using published methods, or purchase it from a nucleic acid synthesis company17,21,49,50,51.
- If inserting a gene, synthesize or purchase a donor DNA template.
- Store the protein and RNA aliquots at -80 °C and thaw on ice immediately before use.
NOTE: Each freeze-thaw slightly lowers the efficiency. Detailed, open-access protocols for Cas9 purification52 and the in vitro transcription of sgRNAs53 are available elsewhere.
- After choosing which gene to target, use one of the free online tools to design an optimal gRNA44,45,46,47,48. Be sure to target an exon if hoping to generate a knockout.
- If working with C. elegans, skip to step 1.5. For the P. hawaiensis protocol, skip to step 1.6. If using sgRNA, skip to step 1.4. Proceed to step 1.3 to assemble a gRNA for primary cell editing.
- Assemble a gRNA by mixing equimolar amounts of tracrRNA and crRNA. Make 100 µL of 80 µM gRNA stock, for about 50 genome editing experiments.
- Incubate the gRNA at 37 °C for 30 min and then allow it to slowly cool to room temperature.
- RNP prep for HSPC and T cell editing: Assemble an RNP complex by mixing a 1 - 2x molar amount of gRNA to 200 pmol of Cas9 protein in a total volume of 10 µL. Very slowly, add concentrated Cas9 to the gRNA (pre-diluted in the Cas9 buffer) for about 30 s, making quick circles with the pipette, bringing the final Cas9 concentration to 20 µM.
- Prepare the electroporation cuvettes.
NOTE: This protocol is specific to the commercial system referred to in the Table of Materials, but RNP editing can also be achieved with other electroporation devices. - Add 5 µL (100 pmols, T cells) or 10 µL (200 pmol, HSPCs) of RNP to each cuvette.
- If inserting new DNA rather than making a knockout, add 1 µL of 100 µM (100 pmol) single-stranded oligonucleotide donor DNA (ssODN)25,54,55 to the cuvettes or wells of the plate.
- Skip to step 2 for the next instructions in the primary cell editing protocol.
- Prepare the electroporation cuvettes.
- RNP prep for C. elegans editing: Assemble the RNP complex by adding the following reagents in order to create a final volume of 20 µL (the final concentrations are noted in parentheses): Cas9 (2 µM), HEPES pH 7.5 (10 µM), KCl (115 µM), crRNA (12 µM), tracrRNA (40 µM), and the repair templates if needed (0.5 µM ssDNA or up to 350 ng/µL dsDNA).
NOTE: The efficiency of a Cas9-mediated DSB-templated repair is proportional to the concentration of the dsDNA repair construct; thus, the higher the concentration of the repair template, the more efficient the templated repair. However, an injection of mixes containing greater than 350 ng/µL of dsDNA has been shown to reduce the viability of the injected worms. Thus, it is best to use up to, but no more than 350 ng/µL of dsDNA in the mix to maximize the repair efficiency while minimizing its lethality.- Add multiple crRNAs to target multiple loci simultaneously, as needed for the co-CRISPR/co-conversion screening approach described in step 5.4. When adding more than one crRNA, add each sequentially to the master mix.
NOTE: The amount of each crRNA does not need to be the same, and even doubling the total concentration of crRNAs in the master mix without changing the concentration of Cas9 does not appear to interfere with the frequency of mutagenesis at a specific locus. Examples are described in detail in Paix et al.56. - Mix by pipetting and spin the RNP solution at 16,000 x g for 5 s to ensure that the solution is collected at the bottom of the tube.
- Incubate the solution at 37 °C for 15 m.
- Centrifuge the sample at 16,000 x g for 1 min to pellet any particulates that could clog the thin-bored microinjection needle. Use the supernatant in the subsequent steps.
- Skip to step 4 for the remainder of the C. elegans protocol.
- Add multiple crRNAs to target multiple loci simultaneously, as needed for the co-CRISPR/co-conversion screening approach described in step 5.4. When adding more than one crRNA, add each sequentially to the master mix.
- RNP prep for P. hawaiensis editing: Prepare single-use Cas9 aliquots by diluting them with nuclease-free water and phenol red (for visualizing injections) to a final concentration of 6.25 µM of Cas9 and 0.15% phenol red.
- Assemble the RNP complex by mixing a 2-5x molar excess of gRNA to the Cas9 protein in a total volume of 6 µl. Add 12 pmol of Cas9 to gRNA, bringing the final Cas9 concentration to 2 µM, gRNA concentration to 4 - 8 µM, and phenol red concentration to 0.05%.
- Incubate the mixture at room temperature for 10 min to complex the RNP.
- Skip to step 6 for the next instructions in the P. hawaiensis editingprotocol.
2. Cell Culture and Preparation
NOTE: Perform steps 2.1.1 to 3.3.3 in a biological safety cabinet.
- Purchase cryopreserved human mobilized peripheral blood CD34+ HSPCs from a vendor.
- Thaw ~1 x106 HSPCs in a 37 °C water bath for 3 min and transfer them to a 15 mL conical tube. Add 10 mL of a serum-free expansion medium from a commercial source and spin the mixture at 100 x g for 10 min. Remove the supernatant and resuspend the cells in 2 mL of supplemented SFEM. Plate the cells in 6-well plates and culture them in a 37 °C incubator for 24-48 h prior to the RNP electroporation.
- Count the cells with a hemocytometer and transfer the total number of HSPCs needed (150,000-200,000 HSPCs per cuvette to be electroporated) to a centrifuge tube.
- Spin the tube at 100 x g for 10 min to pellet the cells.
- Purchase human primary CD4+ T cells from a vendor or isolate them from human whole blood by the density gradient centrifugation29.
- Prior to the T cell activation, pre-coat 48-well culture plates with αCD3 (UCHT1) and αCD28 (CD28.2). Coat the plates with 500 µL of 10 µg/mL αCD3 and 10 µg/mL αCD28 in PBS for at least 2 h at 37 °C.
NOTE: For some loci, NHEJ can be achieved without pre-stimulation, but including this step maximizes its efficiency. - Culture the T cells for 48 h at 37 °C on αCD3/αCD28 antibody-bound plates in a RPMI complete medium [RPMI-1640 supplemented with 5 mM of HEPES, 2 mM of commercial alternative to L-Glutamine, 50 µg/mL of penicillin/streptomycin, 50 µM of 2-mercaptoethanol, 5 mM of non-essential amino acids, 5 mM of sodium pyruvate, and 10% (vol/vol) FBS]. Culture the T cells at a density of 2,000,000 T cells in 500 µL of media per well of a 48-well plate.
- Count the T cells using a hemocytometer and transfer the total number of T cells necessary for the electroporation experiment (100,000-1,000,000 T cells per cuvette to be electroporated) to a centrifuge tube.
- Spin the tube at 90 x g for 8 min to pellet the cells. If the cells have been density gradient-separated within 2 days, spin them at 200 x g for 8 min.
- Prior to the T cell activation, pre-coat 48-well culture plates with αCD3 (UCHT1) and αCD28 (CD28.2). Coat the plates with 500 µL of 10 µg/mL αCD3 and 10 µg/mL αCD28 in PBS for at least 2 h at 37 °C.
- For both cell types, aspirate the supernatant with a pipette/vacuum, removing any bubbles.
- Gently resuspend the cells with 20 µL of electroporation buffer per cuvette.
- Add 20 µL of the cells (150,000-200,000 HSPCs or 100,000-1,000,000 T cells) to each cuvette, which already contains 10 µL of the RNP, and mix well by pipetting up and down without creating bubbles.
3. RNP Electroporation
- Electroporate the cuvettes after placing them in a nucleofector. For the HSPCs, use the pulse code ER100. For the T cells, use the pulse code EH-115.
- HSPCs only: Add 100 µL of a supplemented SFEM medium (warmed to 37 °C) to each cuvette immediately after electroporation and let the cells recover for 10-15 min.
- Transfer the cells to culture them in a 96-well round-bottom plate and add an additional 100 µL of the supplemented SFEM medium for 24 h.
- Change them to a fresh supplemented SFEM medium and incubate them for an additional 24 - 72 h.
- Remove the cells for genotyping them 48-96 h post-electroporation. Spin the cells at 300 x g for 5 min and remove the supernatant before beginning the DNA extraction (step 8.2).
- T cells only: Add 80 µL of RPMI complete culture media pre-warmed to 37 °C from the reservoir to each cuvette or well, using a multi-channel pipette (if necessary).
- Incubate them at 37 °C for 15 min.
- Add the appropriate media, antibodies, cytokines, etc. to the destination plate(s) and pre-warm them in a 37 °C incubator.
- Transfer 107 µL of the electroporated cells from the wells to a round-bottom 96-well plate using a multi-channel pipette (if necessary).
- For information on assessing the editing outcomes, skip to step 8.
4. C. elegans Preparation
- 1 day prior to microinjection: Prepare the agarose pads for the microinjection.
- Make a 3% (w/v) agarose solution in water by adding agarose to water and bringing the solution to a boil on a hot plate or in a microwave.
- Arrange 24 mm x 50 mm x 1.5 mm cover glass slides on a table and use a glass Pasteur pipette to place a small (~15 µL) drop of agarose solution onto the slide. Quickly flatten the agarose drop by placing another coverslip on top. Allow the agarose to solidify and then remove one of the coverslips.
- Leave the agarose-coated coverslip face-up on a tabletop overnight to dry. After 24 h, store the agarose pads in a clean, dry container.
NOTE: These can be used indefinitely.
- Pull the microinjection needles: using borosilicate glass capillaries with filaments (outer diameter 1.0 mm and inner diameter 0.58 mm), pull the needles based on Mello and Fire57 and other resources58. The needles can be used immediately or can be stored in a clean, dry container, braced by clay supports.
- For the maintenance of the worms, prepare a Nematode Growth Media (NGM) agar poured into Petri plates and spotted with OP50 bacteria (for protocols on standard C. elegans maintenance and recipes for growth media, see Stiernagle59).
- Stage the worms for microinjection: 12-24 h prior to the microinjection, pick L4-staged hermaphrodites to a new NG-agar plate with OP50 bacteria and incubate them overnight at 20 °C. For each Cas9 target/injection mix, pick ~30 worms to the plate.
- Day of microinjection: Load the pulled microinjection needle with the RNP solution supernatant prepared in step 1.5.
- Pipette the supernatant from step 1.5.4 into a pulled capillary pipette and backfill the solution from the capillary pipette into the prepared microinjection needle (generally loading less than 0.1 µL).
- Mount the loaded needle onto the microinjection apparatus attached to a micromanipulator. Set the injection apparatus pressure to 250 kPa and the balance pressure to 25 kPa.
- Break back the loaded needle tip to generate a sharp needle edge. Place a 15 mm x 15 mm x 1.5 mm square coverslip on the top of a 24 mm x 50 mm x 1.5 mm coverslip.
- Overlay one edge of the square coverslip with halocarbon oil 700.
- Position the needle in the oil, at the edge of the 15 mm square coverslip.
- Using a hand to guide the microscope stage and coverslip, brush the slide up and along the edge of the needle while depressing the injection pedal/button. Break the needle tip back, increasing the flow of the liquid out of the needle. Achieve an optimal flow rate by making the injection mix flow up along the edge of the needle, forming ~1 bubble/s.
- Confirm that the L4 worms picked 12-24 h prior to microinjection are developmentally staged young adults on the day of injection. Pick the young adult worms to an NG-agar plate that lacks OP50 bacteria and allow them to crawl around for 5 min. This reduces the quantity of bacteria transferred to the injection pad, minimizing needle clogs.
- Place an agarose injection pad/coverslip onto a dissection scope. Using a worm pick, lay a small track of halocarbon oil along one edge of the pad.
- Using the worm pick coated in oil, lift several worms off the NG-agar plate and into the track of oil. With a fine hair attached to a pipette, such as an eyelash or cat whisker, position the worms in parallel, gently pushing the worms into the agarose pad. Until comfortable with the microinjection procedure, only mount and inject one worm at a time.
NOTE: The dry agarose will wick the moisture from the worms, causing them to adhere to the pad. Consequently, one must work quickly as the worms can desiccate.- Once in position and attached to the pad, overlay the worms with another few drops of halocarbon oil (~20 µL) from the tip of the worm pick.
5. C. elegans Gonad Microinjection with RNPs and Post-injection Care
NOTE: The microinjection protocol is adapted from Mello and Fire57and described in detail elsewhere60,61.
- Place the coverslip with the mounted worms onto the injection microscope. Under a low magnification (5X objective, 10X ocular), position the worms perpendicular to the injection needle.
- Switch to a high magnification (40X objective, 10X ocular), reposition the needle adjacent to the gonad arm corresponding to the region near the nuclei in mid- to late-pachytene.
- Using the micromanipulator, move the needle against the worm, depressing the cuticle slightly. Then, with one hand, tap the side of the microscope stage to jolt the needle through the cuticle. Depress the injection pedal/button and slowly fill the gonad arm with the injection mix and remove the needle.
- Repeat this step with the other gonad arm.
- Once the worms are injected, remove the coverslip/agarose pad and place it under a dissecting microscope.
- Using a pulled capillary pipette, displace the oil from the worms by pipetting an M9 buffer over them. Perform this treatment to release the worms from the agar.
- After 10 min, when the worms are thrashing around in the buffer, move them to an NG-agar plate with OP50 bacteria using the pulled capillary pipette. Place the plate at 20 °C for 2-3 h until the worms have recovered and are moving around.
- Once recovered, individually transfer the worms to NG-agar plates with OP50 and transfer the plates to a 25 °C incubator.
- Allow the P0-injected worms to grow and lay progeny for 3 days. Screen the F1 offspring.
- If using co-CRISPR or co-conversion62,63,64,65, then select the candidate worms for screening based on whether they have the mutant phenotype of the reference gene. Individually transfer these marked worms to new NG-agar plates with OP50 and allow them to lay F2 progeny at 20 °C.
NOTE: The phenotype used for a co-CRISPR screening or selection should provide an early estimate for the success of Cas9 editing. - If the co-CRISPR phenotype is not present, microinject a positive control plasmid to assist in improving the microinjection efficiency.
NOTE: For instance, including a plasmid in the injection mix that encodes mCherry-tagged MYO-2 will help assess the injection efficiency. Worms successfully injected with pCFJ90 will have some offspring with fluorescent pharynxes.
- If using co-CRISPR or co-conversion62,63,64,65, then select the candidate worms for screening based on whether they have the mutant phenotype of the reference gene. Individually transfer these marked worms to new NG-agar plates with OP50 and allow them to lay F2 progeny at 20 °C.
- Examine the F1 worms for the presence of the desired edits. Pick the F1 mother to an individual well of a 96-well plate, lyse her, and examine her DNA by either insert-specific PCR amplification, DNA sequence analysis, or surveyor nuclease assay (CEL-1)66.
NOTE: These assays can be performed when using a co-CRISPR/co-conversion or other screening or selection regimes65,66,67,68. - For information on assessing the editing outcomes, skip to step 8.
6. P. hawaiensis Preparation
- 1 day prior to the microinjection, enrich for the early embryos by setting up a 'pair tank' the night before; newly separated females will contain freshly-fertilized embryos. See Rehm et al.69 for details.
- On the day of microinjection, collect the single-cell Parhyale embryos (0-4 h post-fertilization) by anesthetizing gravid females with 0.02% clove oil in seawater and gently scraping the embryos out of her ventral brood pouch using a flame-pulled and rounded glass pipette and a dull pair of #3 forceps.
7. P. hawaiensis Embryo Microinjection with RNPs and Post-injection Care
- Backfill a pulled capillary tube with approximately 1 µL of the RNP injection mix described above.
- Use compressed nitrogen to microinject each embryo as described in Rehm et al.69.
- Inject the Parhyale embryos under a dissecting microscope using a microinjector and a micromanipulator. Load 1.5 µL of the injection mix into the back of a pulled capillary tube (4 inches-1.0 mm with filaments, pulled using a micropipette pulling apparatus) using a microloader pipette tip.
- Set up the needle on the injection apparatus and break the tip of the needle (a very small amount) using a pair of forceps under the dissecting scope. Calibrate the volume delivered by injecting into halocarbon oil 700 and measuring the diameter of the bubble.
- Cut a 'trough' out of the curing agent using a razor blade. Fill it halfway with filter-sterilized sea water, and line the Parhyale embryos up in the trough to stabilize.
- Inject the embryos using the microinjection setup, stabilizing each embryo with a pair of forceps during the injection. After injection, use a glass transfer pipette to transfer the embryos over to a fresh 60 mm culture dish filled halfway with filter-sterilized sea water.
- If the first division has already occurred to form a 2-cell embryo (4-6 h post-fertilization), generate fully-mutant animals by injecting both blastomeres. To ensure a total cleavage of the 2-cell stage, co-inject the blastomeres with FITC or TRITC dextran and observe that the signal is restricted to a single blastomere under a fluorescent dissecting scope after the injection.
- Alternatively, generate 'half-mutant' animals by injecting just one of the two blastomeres at the 2-cell stage (roughly divided left-right depending on the tissue and position along the A-P axis).
- Inject one cell in an 8-cell embryo (7.5-9 h post-fertilization) to restrict the editing to a single germ layer. See Gerberding et al.70 for a map of early blastomere lineages.
- Incubate the embryos in 60 mm culture dishes (no more than 25 per dish), filled halfway with filter-sterilized seawater, 'pre-oxygenated' using an aquarium bubbler or by shaking vigorously.
- Place the dishes of embryos in a loosely-sealed plasticware lined with wet paper towels to maintain humidity and place them in a 26 °C incubator with a 12 h light-dark cycle.
- Transfer the surviving embryos to clean seawater dishes every few days.
NOTE: Embryos may be cultured at room temperature, although they will develop much more slowly.
- Dissect and fix the embryos at various stages for an expression analysis by in situ hybridization or antibody staining (see Browne et al.71 for a staging guide, and additional references for dissection and fixation72, in situ hybridization73, and antibody staining74).
- Make dissection needles by threading a bent piece of tungsten wire approximately 0.5 in in length into the end of an insulin needle. Sharpen the needle in sodium hydroxide under a current. Use a 1 mL syringe as the handle of the dissection needle.
- Fill one well of a 3-well glass dish halfway with a freshly-made solution of 9 parts PEM Buffer (0.1 M of PIPES pH 6.95, 2 mM of EGTA, 1 mM of MgSO4), 1-part 10x PBS, and 1 part 32% PFA. Place 3-5 embryos into the dish and poke a small hole into each embryo, using a sharp tungsten needle to poke and a slightly dulled one to stabilize, allowing the yolk to flow out and the fixative to run in.
- Using a pair of sharpened tungsten needles, gently tease away the outer two membranes surrounding the Parhyale embryo. Dissect them in fixative to make the embryos more robust but work quickly to keep the membrane from becoming fixed to the embryo, which makes membrane removal more difficult. Allow the embryos to fix for a total of 15-20 min. for antibody staining or 40-50 min for in situ hybridization.
- Image live hatchlings and analyze them for morphological and behavioral phenotypes or fix and stain them for more detailed analyses. Raise the hatchlings to sexual maturity in 2-3 months to establish knockout and transgenic lines (see Kontarakis and Pavlopoulos75 for hatchling care and other useful details).
8. Assessing Editing Outcomes
- If applicable, look for a visual or functional phenotype in the edited cells or organisms.
NOTE: This process will vary widely by application, and some examples are described at the end of their relevant protocol steps above. After correcting the sickle cell mutation in HSPCs, analyze the hemoglobin production by differentiated erythroblasts using HPLC (Figure 1A). A knockout of the IL-2 receptor gene in T cells can be confirmed by surface staining and flow cytometry (Figure 1B). To assess C. elegans and P. hawaiensis phenotypes, observe the animal morphology and behavior under a light or fluorescent microscope (Figures 1C and 1D). - To determine the efficiency and type of the genomic edits generated, lyse the pools of edited cells and extract their genomic DNA using a commercial extraction kit21.
- For a quick estimation of indel formation, PCR-amplify at least 200 base pairs around the cut site and perform a T7 endonuclease1 (T7E1)76 or surveyor (CEL-1 nuclease) assay77.
- If an indel formation at the Cas9-cut site or successful HDR will create or remove a known restriction site, consider using a restriction enzyme digestion to estimate the editing efficiency6. The restriction fragment length polymorphism (RFLP) assay can be a convenient way to check the efficiency if it happens to be available.
- For an accurate quantification of the editing efficiency and determination of predominant editing outcomes, send the PCR amplicon for a standard Sanger sequencing with both forward and reverse primers.
NOTE: If analyzing a single clone or organism, the analysis of Sanger results is simple, as demonstrated in Figure 2A. If analyzing a pool of cells, then analyze the chromatograms with the online tool78, as shown in Figure 2B. - For a full quantification and sequences of editing outcomes, perform deep sequencing27,54, as depicted in Figure 2C.
- To assess a particular set of off-target changes, PCR-amplify the predicted off-target sites and send them for NGS. To enable the detection of chromosomal translocations, perform GUIDE-seq79 or high-throughput, genome-wide translocation sequencing (HTGTS)80. For a complete picture of the off-target edits in a clonal population, perform whole-genome sequencing (WGS)81,82,83.
NOTE: There are a variety of methods for quantifying on- and off-target genome edits, explained further in various review articles84,85,86.
Representative Results
These experiments show how pre-assembled Cas9 RNP can be used to manipulate the genomes of primary cells and whole organisms. Researchers purify or purchase Cas9 protein and sgRNA, combine the two components to pre-form the complex, and introduce the RNP into their cells or organism of interest. After allowing enough time for editing to occur and for offspring of the next generation to be born (if applicable), check for phenotypes and/or collect cells for genotyping. Phenotypes may be observed via functional assays, expression assays, visualization (by eye or with microscopy), or other methods, depending on the experiment.
For example, HSPCs that have been edited to correct the β-globin mutation that causes sickle cell disease can be differentiated into erythrocytes and assayed for the production of healthy or sickle hemoglobin27,87 (Figure 1A). T cells edited to knock out the high-affinity IL-2 receptor gene, CD25 (IL2RA), can be analyzed by surface staining and flow cytometry88, and functionally analyzed to detect a signaling response to IL-2 stimulation (Figure 1B). T cells can also be reprogrammed in many clinically important ways that require assessment of different phenotypes, including the efficacy of HIV infection89 and the in vivo antitumor efficacy of CAR-T cells11.
Using a co-CRISPR/co-conversion screening approach, C. elegans worms are edited simultaneously at two loci62. HDR at the dpy-10 reference gene using a ssODN repair template results in an easily-scored dominant dpy-10 gain-of-function mutation. Heterozygous F1dpy-10(gof) animals are roller (Rol) and homozygous dpy-10(gof) animals are dumpy (Dpy). The presence of the phenotype indicates that Cas9 editing occurred in these animals and improves the odds of identifying an editing event at the second locus in the Rol or Dpy F1 animals. A successful editing experiment should result in 33-50% of injected P0 worms yielding 20 or more F1 offspring that are Rol or Dpy90. It is then possible to choose non-Rol animals to return dpy-10 to wildtype and select for the homozygous edit of interest. As a rule of thumb, the concentration of the crRNA targeting the co-CRISPR reference gene should be half that of the crRNA targeting the gene of interest. If an edit in the gene of interest is not recovered, the ratios of the two CRISPR RNAs can be adjusted to increase the likelihood of recovering the desired mutation. For instance, increasing the amount of crRNA for the gene of interest relative to the reference gene crRNA will increase the percentage of worms possessing edits in the gene of interest within the population of worms that also possess edits at the reference gene locus. Co-conversion frequencies vary, but the rates are typically 20-60%, often yielding homozygous edits in the F1 generation (Figure 1C).
P. hawaiensis hatchlings that have been edited to knock out the Abdominal-B gene (Abd-B) display clear morphological abnormalities3 (Figure 1D). This gene is required for correct abdominal patterning, and its disruption results in thoracic-type jumping and walking legs replacing the swimming and anchor legs that are usually present on the abdomen.
Determining genome editing outcomes at the genotypic level requires either sequencing or an in vitro assay that detects sequence changes. Here, we show representative sequencing data from our model cell types and organisms, highlighting different approaches to editing quantification. Note that the figure labels are generalized because all methods shown here can be applied to any biological system.
Sequencing-based approaches vary in technical complexity and depth of results. For clonal edited populations or easily-separable individual organisms, edited individuals can be sequenced following genomic DNA extraction. Standard Sanger sequencing results will reveal the sequence change at the Cas9-cut site in a given individual, with hypothetical frameshifts that would disrupt its function (Figure 2A). The online tool used for sequencing is another Sanger sequencing-based approach that can be applied to mixed populations rather than individual mutants78. Sequences are analyzed with an online tool that can approximate overall editing efficiency as well as predominant sequence outcomes. The representative data are shown in Figure 2B.
The most thorough sequencing method described here is deep sequencing (sometimes referred to as high-throughput or next-generation sequencing). This method provides DNA sequences from individual genomes in a mixed population. Such data can be illustrated in a variety of ways. Here, we have classified individual sequencing reads from edited cells based on the editing outcome (Figure 2C). Most cells are edited via the NHEJ pathway, which typically results in gene disruption. In others, the target gene has been swapped out for an alternate version via HDR27.
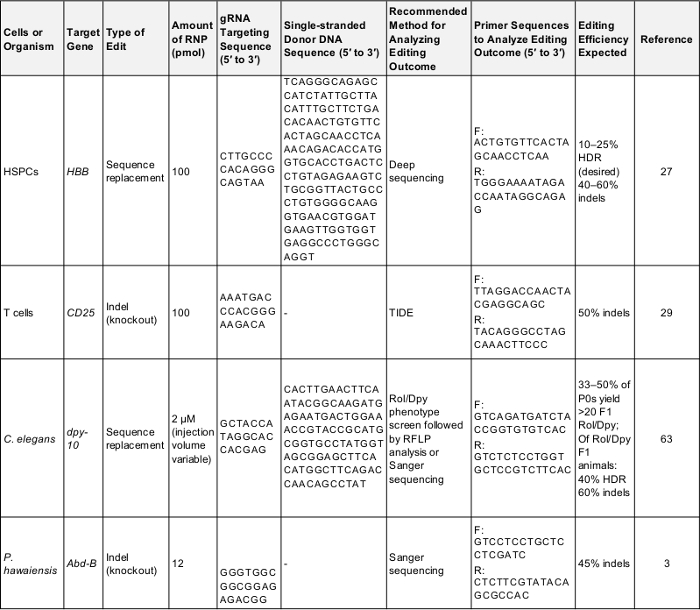
Table 1: Positive controls for preliminary genome editing experiments. This table shows the key information needed to perform a first-time genome editing experiment in each of the cells and organisms described in this protocol. Following these parameters is likely to yield a successful result that can be used to test the protocol or as a baseline for comparison once the experimenter is targeting a gene of their own interest. F: forward, R: reverse, HDR: homology-directed repair. Please click here to download this table.

Figure 1: Representative phenotypic results from Cas9 RNP editing of primary human cells and organisms. (A) This is an HPLC trace showing that after successful genome editing, HSPCs that are differentiated into late-stage erythroblasts will produce more functional hemoglobin than sickle hemoglobin. Mutant erythrocytes produce sickle hemoglobin (HbS), while successfully-edited cells will produce healthy hemoglobin (HbA and HbA2) as well as fetal hemoglobin (HbF). The absorbance is graphed in arbitrary units (au). This panel was first published in DeWitt et al.27. It is reprinted with permission from the American Association for the Advancement of Science. (B) On the left, for each condition, this panel shows flow cytometry data showing that surface-stained T cells do not express CD25 after the CD25 gene has been knocked out with RNP. The CD25 abundance is plotted on the x-axis with the cell size on the y-axis. On the right, for each condition, this panel shows the Phospho-Stat5 (pStat5) quantification after an induction with IL-2. The signaling is reduced when the IL-2 receptor is absent (CD25 KO). The pStat5 abundance is plotted on the x-axis and the data resulting from three different levels of IL-2 input are compared vertically. (C) This panel shows a Caenorhabditis elegans co-CRISPR/co-conversion screen targeting dpy-10 as the co-conversion marker. Two guide RNAs target two loci, dpy-10 and your favorite gene (yfg), in the same P0-injected animal. HDR at dpy-10 results a Rol or Dpy phenotype. The selection of Rol- or Dpy-F1 animals increases the chances of identifying edits at the second locus. (D) This panel shows that wildtype Parhyale hawaiensis hatchlings have normal abdomens with swimming and anchor legs. The Abd-B knock-out hatchlings (F0 individuals) develop an abdomen transformed towards thorax. Thus, the swimming and anchors legs are gone and replaced by the jumping and walking legs associated with a normal thorax. Please click here to view a larger version of this figure.

Figure 2: Typical results from editing outcome analysis methods. (A) This panel shows examples of the Sanger sequencing results from individual F1 P. hawaiensis organisms, including the wildtype sequence and three different indels that disrupt the gene function by shifting the open reading frame. (B) These TIDE results show the range of insertions and deletion events that occurred at a Cas9-target site in a pool of sequenced T cells. The x-axis indicates the length of a given insertion or deletion in nucleotides. (C) These deep sequencing results show no genome editing without nucleofection or gRNA, and successful editing with intact Cas9 RNP, grouped by DNA repair outcome in HSPCs. Please click here to view a larger version of this figure.
Discussion
Establishing a robust genome editing protocol in a cell line or organism of interest requires the optimization and empirical testing of several key parameters, discussed in this section. Trying a few variations of the general approaches presented here is highly encouraged. The key limitation of this protocol is that applying these methods to other cells or organisms may lead to a different outcome depending on the species studied, and an experimental design that leads to a high-efficiency gene knockout may not promote DNA insertion. Thus, we recommend starting with the methods presented here and troubleshooting as described below.
Troubleshooting genome editing reagent quality:
Generating or purchasing high-quality reagents is a critical step in any genome editing protocol. Cas9 protein can be purified in the lab or purchased commercially. Many protocols note a final concentration for Cas9 in RNP recipes, but the optimal gene editing activity will depend on the specific activity of any individual Cas9 protein preparation, which varies depending on the source. Once the protocol presented here is working, consider optimizing the amount of RNP used by titrating Cas9 levels to establish an optimal concentration: one that provides highly specific target DNA cleavage without unnecessary off-target cleavage caused by excessive Cas940.
Guide RNA purity and homogeneity can also be determinants of genome editing success22. Purchased sgRNAs or separate crRNA and tracrRNA components are generally high-quality reagents and a variety of chemical modifications are available to combat problems with RNA degradation or to imbue additional features to the RNP91. While chemically-modified gRNAs may not be necessary for standard genome editing experiments, some groups have observed much higher editing efficiencies with such reagents, so they may be worth trying after mastering the process and/or when gRNA degradation appears to be an issue22,91. In vitro transcription and subsequent gel purification is an inexpensive alternative, which may be sufficient for routine genome editing experiments17,21,49,50. Further, several approaches that are commonly applied to produce homogenous gRNA populations in vivo, including ribozyme- and tRNA-based excision of individual guides, may be extended to in vitro RNA preparation to generate cleaner products92.
Guide RNA and donor DNA design tips:
Guide RNA selection is a critical factor in achieving highly efficient on-target editing while minimizing the chances of off-target cleavage. To aid in guide selection, several studies have used high-throughput screens coupled with next-generation sequencing to compile sequence features of successful guides47,79,93,94,95,96. These features have been used to develop predictive algorithms and online tools to assist in guide selection44,45,46,47,48. Such algorithms are grounded on screens using DNA-based systems for guide RNA expression. Guides are expressed using a Pol III promoter, and their expression is therefore prone to the limitations associated with Pol III transcription, such as premature termination when encountering tracks of uracil97,98,99. However, use of RNPs made with in vitro-synthesized guide RNAs bypasses those concerns and simplifies the constraints on guide design. A common feature that emerged from these algorithms and has been confirmed in numerous studies with highly effective genome editing, is the presence of a purine, particularly a guanine, at the 3′ end of the guide's target-specific sequence. This guide feature has been very successful among organisms ranging from mammals to C. elegans, fruit flies, and zebrafish65,100,101. In addition, for C. elegans, designing guides with a GG dinucleotide at the 3′ end of the guide's targeting region is an effective strategy for predicting highly effective guide RNAs65. Ideally, test multiple guides in parallel to determine which is most successful for a given application.
When attempting to introduce a DNA sequence into the genome, the design of the donor or template DNA is also crucial. Single-stranded oligonucleotide donors (ssODNs) are inserted more reliably than other typical repair templates, linear double-stranded and plasmid DNA54,55,102. At some loci, HDR efficiency can be improved with ssODNs that are complementary to the non-target or displaced DNA strand and possess homology arms that are asymmetric in length27,55. Since the repair template is being inserted at the cut site and includes the targeted sequence, steps must be taken to prevent Cas9 from cleaving the donor DNA before or after the genomic insertion. This is accomplished by making silent mutations to the PAM sequence or seed region, avoiding the recognition by Cas9 while retaining the function of the inserted gene21,103. Though even single nucleotide changes to the PAM are likely to abolish binding104, try to change at least four nucleotides to be safe.
Significance and future applications:
Genome editing with CRISPR-Cas9 has emerged as a powerful method enabling facile genetic manipulation of any organism. Editing with the Cas9 RNP takes a bit more effort at first but is straightforward to use once reagents and protocols are established in a lab. Editing cells with pre-assembled RNP instead of plasmid DNA leads to higher overall editing efficiencies, including the difficult-to-achieve gene insertion via HDR, with fewer off-target effects24,25,26,27,29. Further, experimenters avoid problems with gene expression, RNA degradation, protein folding, and the association between gRNA and Cas9 molecules synthesized separately within the cell22,23. RNP editing also circumvents safety concerns about insertional mutagenesis and sustained expression that may arise when viral delivery methods are used clinically14. Because of these advantages, many scientists conducting pre-clinical, proof-of-concept experiments favor RNP editing for human therapeutic applications. Both in vivo and ex vivo RNP-based genome editing approaches are in development to treat or even cure a variety of conditions, from genetic diseases like Duchenne muscular dystrophy105 and sickle cell disease27 to HIV29 and cancer11. Interestingly, Cas9 RNP is increasingly employed as a delivery method for agricultural engineering because it enables 'DNA-free' editing of plants33,34,36.
Disclosures
The authors Alexander Marson and Jacob E. Corn are co-founders of Spotlight Therapeutics. Jacob E. Corn is an advisor to Mission Therapeutics and his laboratory has received sponsored research support from AstraZeneca and Pfizer. Alexander Marson is an advisor to Juno Therapeutics and PACT Therapeutics, and his laboratory has received sponsored research support from Juno Therapeutics, Epinomics, and Sanofi. His laboratory has also applied for patents related to Cas9 RNP technology.
Acknowledgments
We thank many previous members of our labs and the Bay Area genome editing community for their contributions to the development of these methods. We thank Ross Wilson for critically reading this manuscript.
Alexander Marson's research is supported by a gift from the Jake Aronov and a National Multiple Sclerosis Society grant (CA 1074-A-21). Alexander Marson holds a Career Award for Medical Scientists from the Burroughs Wellcome Fund and is a Chan Zuckerberg Biohub Investigator. Jacob E. Corn's research is supported by the Li Ka Shing Foundation, the Heritage Medical Research Medical Institute, and the California Institute for Regenerative Medicine. Behnom Farboud and Barbara J. Meyer's research is funded in part by the NIGMS grant R01 GM030702 to Barbara J. Meyer, who is an investigator of the Howard Hughes Medical Institute. Erin Jarvis and Nipam H. Patel's research is funded in part by the NSF grant IOS-1257379 and Erin Jarvis acknowledges support from an NSF GRFP and a Philomathia Graduate Fellowship.
Materials
| Name | Company | Catalog Number | Comments |
| Reagents/Materials | |||
| DNA oligonucleotides | Integrated DNA Technologies | - | IDT will provide custom DNA sequences, including those in Table 1 |
| Guide RNAs | Synthego | - | Synthego will provide high-quality sgRNAs for S. pyogenes Cas9, including custom sgRNAs containing the targeting sequences included in Table 1 |
| Purified Cas9 protein (EnGen Cas9 NLS, S. pyogenes) | New England Biosciences | M0646T | If possible, purifying Cas9 in-house or purchasing from local core facilities is a less expensive option |
| Normal peripheral blood CD34+ stem/progenitor cells | AllCells | PB032-2 | |
| StemSpan SFEM | StemCell Technologies | 09650 | |
| StemSpan CC110 | StemCell Technologies | 02697 | |
| P3 Primary Cell 4D-Nucleofector X Kit | Lonza | V4XP-3032 | |
| RPMI-1640 Medium, With sodium bicarbonate, without L-glutamine, liquid | Sigma | R0883-6X500ML | |
| EasySep™ Human T Cell Isolation Kit | Stemcell | 17951 | |
| cell culture plate, 96 wells, round | Fisher Scientific | 3799 | |
| CTS (Cell Therapy Systems) Dynabeads CD3/CD28 | Life Tech | 40203D | |
| Reombinant Human IL-2 | UCSF Pharmacy | NA | |
| SepMate-50 500-pack IVD | Stemcell Technologies | 85460 | |
| OP50 Escherichia coli | Caenorhabditis Genetics Center | OP-50 | https://cgc.umn.edu/ |
| Nematode Growth Media agar in petri dishes | - | - | See Stiernagle, T (ref. 59) |
| Standard borosilicate glass capillaries with filament: 4 in (100 mm), 1/0.58 OD/ID | World Precision Instruments | 1B100F-4 | |
| Single-barrel standard borosilicate glass capillaries: 6 in (152 mm), 2/1.12 OD/ID | World Precision Instruments | 1B200-6 | |
| Cover glass; 24 × 50 mm | Thermo Fisher Scientific | 12-544E | |
| Cover glass; 22 × 22 mm | Thermo Fisher Scientific | 12-518-105K | |
| Apex LE agarose | Genesee Scientific | 20-102 | |
| Halocarbon oil 700 | Sigma-Aldrich | H8898-100ML | |
| pCFJ90 plasmid | Addgene | 19327 | |
| Compressed nitrogen | - | ||
| 60 mM culture dishes | BD | ||
| Capillary tubes with filament: 4 in (1.0 mm) | World Precision Instruments | T2100F-4 | |
| Sylgard 184 | Dow Corning | ||
| Petri dishes (100 × 15 mm) | - | ||
| Tungsten wire (0.005 in. diameter) | Ted Pella | ||
| Perfluoroalkoxy alkane (PFA) | - | ||
| Marine salt | - | ||
| 9" pasteur pipettes | - | ||
| Phenol red | - | ||
| Nuclease-free water | - | ||
| Equipment | |||
| 4D Nucleofector | Lonza | AAF-1002X | |
| MZ75 Stereomicroscope | Leica | Out-of-production. Current model is the M80 Stereomicroscope | |
| Axio Vert35 inverted phase contrast fluorescent microscope | Zeiss | Out-of-production. Current model is the Axio VertA.1 | |
| Laser-based micropipette puller (for C. elegans protocol) | Sutter Instrument | FG-P2000 | |
| Picoliter Microinjector (for C. elegans protocol) | Warner Instruments | PLI-100A | |
| Three-axis Joystick oil hydraulic micromanipulator | Narishige International | MO-202U | |
| Coarse manipulator | Narishige International | MMN-1 | |
| Micropipette puller (for P. hawaiensis protocol) | Sutter Instrument | P-80/PC | |
| Microinjector (for P. hawaiensis protocol) | Narishige | IM300 | |
| Microloader pipette tips | Eppendorf | 5242956003 | |
| NG-agar |
References
- Komor, A. C., Badran, A. H., Liu, D. R. CRISPR-based technologies for the manipulation of eukaryotic genomes. Cell. , 1-17 (2016).
- Barrangou, R., Horvath, P. A decade of discovery: CRISPR functions and applications. Nature Microbiology. 2, 1-9 (2017).
- Martin, A., Serano, J. M., et al. CRISPR/Cas9 mutagenesis reveals versatile roles of Hox genes in crustacean limb specification and evolution. Current Biology. 26 (1), 14-26 (2016).
- Goldstein, B., King, N. The future of cell biology: emerging model organisms. Trends in Cell Biology. 26 (11), 818-824 (2016).
- Mali, P., Yang, L., et al. RNA-guided human genome engineering via Cas9. Science. 339 (6121), 823-826 (2013).
- Cong, L., Ran, F. A., et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 339 (6121), 819-823 (2013).
- Deltcheva, E., Chylinski, K., et al. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature. 471 (7340), 602-607 (2011).
- Jinek, M., Chylinski, K., Fonfara, I., Hauer, M., Doudna, J. A., Charpentier, E. A Programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 337 (6096), 816-821 (2012).
- Nowak, C. M., Lawson, S., Zerez, M., Bleris, L. Guide RNA engineering for versatile Cas9 functionality. Nucleic Acids Research. 44 (20), 9555-9564 (2016).
- Jiang, W., Cox, D., Zhang, F., Bikard, D., Marraffini, L. A. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nature Biotechnology. , 1-9 (2013).
- Rupp, L. J., Schumann, K., et al. CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Scientific Reports. 7 (1), 737 (2017).
- Graham, D. B., Root, D. E. Resources for the design of CRISPR gene editing experiments. Genome Biology. 16, 260 (2015).
- Wang, H., La Russa, M., Qi, L. S. CRISPR/Cas9 in genome editing and beyond. Annual Review of Biochemistry. 85, 2270 (2016).
- Nelson, C. E., Gersbach, C. A. Engineering delivery vehicles for genome editing. Annual Review of Chemical and Biomolecular Engineering. 7, 637-662 (2016).
- Yin, H., Kauffman, K. J., Anderson, D. G. Delivery technologies for genome editing. Nature Reviews Drug Discovery. 16 (6), 387-399 (2017).
- Zuris, J. A., Thompson, D. B., et al. Cationic lipid-mediated delivery of proteins enables efficient protein-based genome editing in vitro and in vivo. Nature Biotechnology. 33 (1), 73-80 (2015).
- Cho, S. W., Lee, J., Carroll, D., Kim, J. -S., Lee, J. Heritable gene knockout in Caenorhabditis elegans by direct injection of Cas9-sgRNA ribonucleoproteins. Genetics. 195 (3), 1177-1180 (2013).
- Wang, W., Kutny, P. M., et al. Delivery of Cas9 protein into mouse zygotes through a series of electroporation dramatically increases the efficiency of model creation. Journal of Genetics and Genomics. 43 (5), 319-327 (2016).
- Chen, S., Lee, B., Lee, A. Y. -F., Modzelewski, A. J., He, L. Highly efficient mouse genome editing by CRISPR ribonucleoprotein electroporation of zygotes. Journal of Biological Chemistry. 291 (28), 14457-14467 (2016).
- Remy, S., Chenouard, V., et al. Generation of gene-edited rats by delivery of CRISPR/Cas9 protein and donor DNA into intact zygotes using electroporation. Scientific Reports. 7 (1), 16554 (2017).
- DeWitt, M. A., Corn, J. E., Carroll, D. Genome editing via delivery of Cas9 ribonucleoprotein. Methods. , 1-7 (2017).
- Hendel, A., Bak, R. O., et al. Chemically modified guide RNAs enhance CRISPR-Cas genome editing in human primary cells. Nature Biotechnology. 33 (9), 985-989 (2015).
- Thyme, S. B., Akhmetova, L., Montague, T. G., Valen, E., Schier, A. F. Internal guide RNA interactions interfere with Cas9-mediated cleavage. Nature Communications. 7, 11750 (2016).
- Kim, S., Kim, D., Cho, S. W., Kim, J., Kim, J. -S. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Research. 24 (6), 1012-1019 (2014).
- Lin, S., Staahl, B. T., Alla, R. K., Doudna, J. A. Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. eLife. 3, 04766 (2014).
- Liang, X., Potter, J., et al. Rapid and highly efficient mammalian cell engineering via Cas9 protein transfection. Journal of Biotechnology. 208, 44-53 (2015).
- DeWitt, M. A., Magis, W., Bray, N. L., Wang, T. Selection-free genome editing of the sickle mutation in human adult hematopoietic stem/progenitor cells. Science Translational Medicine. 8 (360), (2016).
- Ramakrishna, S., Kwaku Dad, A. -B., Beloor, J., Gopalappa, R., Lee, S. -K., Kim, H. Gene disruption by cell-penetrating peptide-mediated delivery of Cas9 protein and guide RNA. Genome Research. 24 (6), 1020-1027 (2014).
- Schumann, K., Lin, S., et al. Generation of knock-in primary human T cells using Cas9 ribonucleoproteins. Proceedings of the National Academy of Sciences of the United States of America. 112 (33), 10437-10442 (2015).
- Lee, J. -S., Kwak, S. -J., et al. RNA-guided genome editing in Drosophila with the purified Cas9 protein. G3: Genes, Genomes, Genetics (Bethesda, MD). 4 (7), 1291-1295 (2014).
- Sung, Y. H., Kim, J. M., et al. Highly efficient gene knockout in mice and zebrafish with RNA-guided endonucleases. Genome Research. 24 (1), 125-131 (2014).
- Menoret, S., De Cian, A., et al. Homology-directed repair in rodent zygotes using Cas9 and TALEN engineered proteins. Scientific Reports. 5, 14410 (2015).
- Woo, J. W., Kim, J., et al. DNA-free genome editing in plants with preassembled CRISPR-Cas9 ribonucleoproteins. Nature Biotechnology. 33 (11), 1162-1164 (2015).
- Malnoy, M., Viola, R., et al. DNA-free genetically edited grapevine and apple protoplast using CRISPR/Cas9 ribonucleoproteins. Frontiers in Plant Science. 7, 1904 (2016).
- Svitashev, S., Schwartz, C., Lenderts, B., Young, J. K., Mark Cigan, A. Genome editing in maize directed by CRISPR-Cas9 ribonucleoprotein complexes. Nature Communications. 7, 13274 (2016).
- Liang, Z., Chen, K., et al. Efficient DNA-free genome editing of bread wheat using CRISPR/Cas9 ribonucleoprotein complexes. Nature Communications. 8, 14261 (2017).
- Shin, S. -E., Lim, J. -M., et al. CRISPR/Cas9-induced knockout and knock-in mutations in Chlamydomonas reinhardtii. Scientific Reports. 6, 27810 (2016).
- Pohl, C., Kiel, J. A. K. W., Driessen, A. J. M., Bovenberg, R. A. L., Nygård, Y. CRISPR/Cas9 based genome editing of Penicillium chrysogenum. ACS Synthetic Biology. 5 (7), 754-764 (2016).
- Grahl, N., Demers, E. G., Crocker, A. W., Hogan, D. A. Use of RNA-protein complexes for genome editing in non-albicans Candida species. mSphere. 2 (3), (2017).
- Rivera-Torres, N., Kmiec, E. B. A standard methodology to examine on-site mutagenicity as a function of point mutation repair catalyzed by CRISPR/Cas9 and ssODN in human cells. Journal of Visualized Experiments. (126), (2017).
- Nandal, A., Mallon, B., Telugu, B. P. Efficient generation and editing of feeder-free IPSCs from human pancreatic cells using the CRISPR-Cas9 system. Journal of Visualized Experiments. (129), (2017).
- Mohr, S. E., Hu, Y., Ewen-Campen, B., Housden, B. E., Viswanatha, R., Perrimon, N. CRISPR guide RNA design for research applications. The FEBS Journal. 283 (17), 3232-3238 (2016).
- Bauer, D. E., Canver, M. C., Orkin, S. H. Generation of genomic deletions in mammalian cell lines via CRISPR/Cas9. Journal of Visualized Experiments. (95), e52118 (2015).
- Hsu, P. D., Scott, D. A., et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nature Biotechnology. 31 (9), 827-832 (2013).
- Heigwer, F., Kerr, G., Boutros, M. E-CRISP: fast CRISPR target site identification. Nature Methods. 11 (2), 122-123 (2014).
- Moreno-Mateos, M. A., Vejnar, C. E., et al. CRISPRscan: designing highly efficient sgRNAs for CRISPR-Cas9 targeting in vivo. Nature Methods. 12 (10), 982-988 (2015).
- Labun, K., Montague, T. G., Gagnon, J. A., Thyme, S. B., Valen, E. CHOPCHOP v2: a web tool for the next generation of CRISPR genome engineering. Nucleic Acids Research. 44, 272-276 (2016).
- Haeussler, M., Schönig, K., et al. Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biology. 17 (1), 148 (2016).
- Lo, T. -W., Pickle, C. S., et al. Precise and heritable genome editing in evolutionarily diverse nematodes using TALENs and CRISPR/Cas9 to engineer insertions and deletions. Genetics. 195 (2), 331-348 (2013).
- Bassett, A., Liu, J. -L. CRISPR/Cas9 mediated genome engineering in Drosophila. Methods. 69 (2), 128-136 (2014).
- Prior, H., Jawad, A. K., MacConnachie, L., Beg, A. A. Highly efficient, rapid and co-CRISPR independent genome editing in Caenorhabditis elegans. G3: Genes, Genomes, Genetics. , Bethesda, MD. (2017).
- Hirsh, A. Cas9 expression and purification protocol. protocols.io. , (2017).
- DeWitt, M. A., Wong, J. In vitro transcription of guide RNAs. protocols.io. , (2017).
- Yang, L., Guell, M., et al. Optimization of scarless human stem cell genome editing. Nucleic Acids Research. 41 (19), 9049-9061 (2013).
- Richardson, C. D., Ray, G. J., DeWitt, M. A., Curie, G. L., Corn, J. E. Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA. Nature Biotechnology. 34 (3), 339-344 (2016).
- Paix, A., Folkmann, A., Seydoux, G. Precision genome editing using CRISPR-Cas9 and linear repair templates in C. elegans. Methods. 121-122, 86-93 (2017).
- Mello, C., Fire, A. DNA transformation. Methods in Cell Biology. 48, 451-482 (1995).
- Sutter Pipette Cookbook. , Available from: https://www.sutter.com/PDFs/pipette_cookbook.pdf (2017).
- Stiernagle, T. Maintenance of C. elegans. WormBook: the online review of C. elegans biology. , (2006).
- Evans, T. C. Transformation and microinjection. WormBook: the online review of C. elegans biology. , (2006).
- Berkowitz, L. A., Knight, A. L., Caldwell, G. A., Caldwell, K. A. Generation of stable transgenic C. elegans using microinjection. Journal of Visualized Experiments. (18), (2008).
- Kim, H., Ishidate, T., et al. A co-CRISPR strategy for efficient genome editing in Caenorhabditis elegans. Genetics. 197 (4), 1069-1080 (2014).
- Arribere, J. A., Bell, R. T., Fu, B. X. H., Artiles, K. L., Hartman, P. S., Fire, A. Z. Efficient marker-free recovery of custom genetic modifications with CRISPR/Cas9 in Caenorhabditis elegans. Genetics. 198 (3), 837-846 (2014).
- Ward, J. D. Rapid and precise engineering of the Caenorhabditis elegans genome with lethal mutation co-conversion and inactivation of NHEJ repair. Genetics. 199 (2), 363-377 (2015).
- Farboud, B., Meyer, B. J. Dramatic enhancement of genome editing by CRISPR/Cas9 through improved guide RNA design. Genetics. 199 (4), 959-971 (2015).
- Wood, A. J., Lo, T. -W., et al. Targeted genome editing across species using ZFNs and TALENs. Science. 333 (6040), 307 (2011).
- Friedland, A. E., Tzur, Y. B., Esvelt, K. M., Colaiácovo, M. P., Church, G. M., Calarco, J. A. Heritable genome editing in C. elegans via a CRISPR-Cas9 system. Nature Methods. 10 (8), 741-743 (2013).
- Dickinson, D. J., Ward, J. D., Reiner, D. J., Goldstein, B. Engineering the Caenorhabditis elegans genome using Cas9-triggered homologous recombination. Nature Methods. 10 (10), 1028-1034 (2013).
- Rehm, E. J., Hannibal, R. L., Chaw, R. C., Vargas-Vila, M. A., Patel, N. H. Injection of Parhyale hawaiensis blastomeres with fluorescently labeled tracers. Cold Spring Harbor Protocols. 2009 (1), (2009).
- Gerberding, M., Browne, W. E., Patel, N. H. Cell lineage analysis of the amphipod crustacean Parhyale hawaiensis reveals an early restriction of cell fates. Development (Cambridge, England). 129 (24), 5789-5801 (2002).
- Browne, W. E., Price, A. L., Gerberding, M., Patel, N. H. Stages of embryonic development in the amphipod crustacean, Parhyale hawaiensis. Genesis. 42 (3), New York, NY. 124-149 (2005).
- Rehm, E. J., Hannibal, R. L., Chaw, R. C., Vargas-Vila, M. A., Patel, N. H. Fixation and dissection of Parhyale hawaiensis embryos. Cold Spring Harbor Protocols. 2009 (1), (2009).
- Rehm, E. J., Hannibal, R. L., Chaw, R. C., Vargas-Vila, M. A., Patel, N. H. In situ hybridization of labeled RNA probes to fixed Parhyale hawaiensis embryos. Cold Spring Harbor Protocols. 2009 (1), (2009).
- Rehm, E. J., Hannibal, R. L., Chaw, R. C., Vargas-Vila, M. A., Patel, N. H. Antibody staining of Parhyale hawaiensis embryos. Cold Spring Harbor Protocols. 2009 (1), (2009).
- Kontarakis, Z., Pavlopoulos, A. Transgenesis in non-model organisms: the case of Parhyale. Methods in Molecular Biology. 1196, Clifton, NJ. 145-181 (2014).
- Kim, H. J., Lee, H. J., Kim, H., Cho, S. W., Kim, J. -S. Targeted genome editing in human cells with zinc finger nucleases constructed via modular assembly. Genome Research. 19 (7), 1279-1288 (2009).
- Qiu, P., Shandilya, H., D'Alessio, J. M., O'Connor, K., Durocher, J., Gerard, G. F. Mutation detection using Surveyor nuclease. BioTechniques. 36 (4), 702-707 (2004).
- Brinkman, E. K., Chen, T., Amendola, M., van Steensel, B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Research. 42 (22), 168 (2014).
- Tsai, S. Q., Zheng, Z., et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nature Biotechnology. 33 (2), 187-197 (2015).
- Frock, R. L., Hu, J., Meyers, R. M., Ho, Y. -J., Kii, E., Alt, F. W. Genome-wide detection of DNA double-stranded breaks induced by engineered nucleases. Nature Biotechnology. 33 (2), 179-186 (2015).
- Smith, C., Gore, A., et al. Whole-genome sequencing analysis reveals high specificity of CRISPR/Cas9 and TALEN-based genome editing in human iPSCs. Cell Stem Cell. 15 (1), 12-13 (2014).
- Veres, A., Gosis, B. S., et al. Low incidence of off-target mutations in individual CRISPR-Cas9 and TALEN targeted human stem cell clones detected by whole-genome sequencing. Cell Stem Cell. 15 (1), 27-30 (2014).
- Kim, D., Bae, S., et al. Digenome-seq: genome-wide profiling of CRISPR-Cas9 off-target effects in human cells. Nature Methods. 12 (3), 237-243 (2015).
- Hendel, A., Fine, E. J., Bao, G., Porteus, M. H. Quantifying on- and off-target genome editing. Trends in Biotechnology. 33 (2), 132-140 (2015).
- O'Geen, H., Yu, A. S., Segal, D. J. How specific is CRISPR/Cas9 really. Current Opinion in Chemical Biology. 29, 72-78 (2015).
- Tsai, S. Q., Joung, J. K. Defining and improving the genome-wide specificities of CRISPR-Cas9 nucleases. Nature Reviews Genetics. 17 (5), 300-312 (2016).
- Hoban, M. D., Cost, G. J., et al. Correction of the sickle cell disease mutation in human hematopoietic stem/progenitor cells. Blood. 125 (17), 2597-2604 (2015).
- Simeonov, D. R., Gowen, B. G., et al. Discovery of stimulation-responsive immune enhancers with CRISPR activation. Nature. , (2017).
- Hultquist, J. F., Schumann, K., et al. A Cas9 ribonucleoprotein platform for functional genetic studies of HIV-host interactions in primary human T cells. Cell Reports. 17 (5), 1438-1452 (2016).
- Paix, A., Wang, Y., et al. Scalable and versatile genome editing using linear DNAs with microhomology to Cas9 sites in Caenorhabditis elegans. Genetics. 198 (4), 1347-1356 (2014).
- Lee, K., Mackley, V. A., et al. Synthetically modified guide RNA and donor DNA are a versatile platform for CRISPR-Cas9 engineering. eLife. 6, (2017).
- Minkenberg, B., Wheatley, M., Yang, Y. CRISPR/Cas9-enabled multiplex genome editing and its application. Progress in Molecular Biology and Translational Science. 149, 111-132 (2017).
- Doench, J. G., Fusi, N., et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nature Biotechnology. 34 (2), 184-191 (2016).
- Doench, J. G., Hartenian, E., et al. Rational design of highly active sgRNAs for CRISPR-Cas9-mediated gene inactivation. Nature Biotechnology. , 1-8 (2014).
- Liu, H., Wei, Z., Dominguez, A., Li, Y., Wang, X., Qi, L. S. CRISPR-ERA: a comprehensive design tool for CRISPR-mediated gene editing, repression and activation. Bioinformatics (Oxford, England). 31 (22), 3676-3678 (2015).
- Wu, X., Scott, D. A., et al. Genome-wide binding of the CRISPR endonuclease Cas9 in mammalian cells. Nature Biotechnology. 32 (7), 670-676 (2014).
- Bogenhagen, D. F., Brown, D. D. Nucleotide sequences in Xenopus 5S DNA required for transcription termination. Cell. 24 (1), 261-270 (1981).
- Cozzarelli, N. R., Gerrard, S. P., Schlissel, M., Brown, D. D., Bogenhagen, D. F. Purified RNA polymerase III accurately and efficiently terminates transcription of 5S RNA genes. Cell. 34 (3), 829-835 (1983).
- Chen, B., Gilbert, L. A., et al. Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell. 155 (7), 1479-1491 (2013).
- Gagnon, J. A., Valen, E., et al. Efficient mutagenesis by Cas9 protein-mediated oligonucleotide insertion and large-scale assessment of single-guide RNAs. PLoS ONE. 9 (5), 98186 (2014).
- Ren, X., Yang, Z., et al. Enhanced specificity and efficiency of the CRISPR/Cas9 system with optimized sgRNA parameters in Drosophila. Cell Reports. 9 (3), 1151-1162 (2014).
- Ran, F. A., Hsu, P. D., Wright, J., Agarwala, V., Scott, D. A., Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nature Protocols. 8 (11), 2281-2308 (2013).
- Serano, J. M., Martin, A., et al. Comprehensive analysis of Hox gene expression in the amphipod crustacean Parhyale hawaiensis. Developmental Biology. 409 (1), 297-309 (2016).
- Sternberg, S. H., Redding, S., Jinek, M., Greene, E. C., Doudna, J. A. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature. , 1-17 (2014).
- Lee, K., Conboy, M., et al. Nanoparticle delivery of Cas9 ribonucleoprotein and donor DNA in vivo induces homology-directed DNA repair. Nature Biomedical Engineering. 1 (11), 889-901 (2017).
