

Summary
組立 Cas9 を利用した複雑なリボ核タンパク質 (RNP) は正確で効率的なゲノムの編集のための強力な方法です。ひと初代細胞や両方の古典的なモデル有機体の出現など、ここでは、私たちは細胞や生物の広い範囲にわたってその有用性を強調します。
Abstract
サイト固有の真核生物ゲノム編集 CRISPR (クラスター化された空間定期的に短い回文繰り返されます)-Cas (CRISPR 関連) システムすぐにさまざまな生物学的質問を追求する研究者の間で当たり前となっています。ユーザーはほとんどの場合簡単にプログラムし直されたガイド RNA (gRNA) との複合体で化膿連鎖球菌由来 Cas9 タンパク質を採用しています。これらのコンポーネントは、細胞に導入され、二本鎖 DNA (dsDNA) ゲノムの相補的な領域とのペアリング ベースで酵素は二重鎖切断 (DSB) を生成する両方のストランドを裂きます。後続の修理は、ランダムな挿入または削除イベント (オクターリピート) または休憩のサイトで実験者によって提供される DNA の取り込みに します。
浄化された単一ガイド RNA と Cas9 蛋白質、形成、RNP に実装済み、細胞に直接配信の使用は、高効率遺伝子編集を実現する強力なアプローチです。遺伝子挿入、多くの場合達成するために挑戦する結果の率を高める特に RNP 編集します。プラスミドを介して配信と比較して、細胞内 Cas9 RNP の短残光は少ないオフのターゲット イベントに します。
その利点にもかかわらず CRISPR 遺伝子編集の多くのカジュアルなユーザーは、このテクニックに不慣れな。エントリへの障壁を下げるための明確な利点と多様なアプリケーションを強調表示のコンテキストの範囲で RNP 戦略を実装するための詳しいプロトコルを概説します。ひと初代細胞、T 細胞と造血幹細胞/前駆細胞 (HSPCs) の 2 種類の編集を取り上げます。またどのように編集可能 Cas9 RNP 最近クラシック モデル回虫線虫などを含む全体の生物の安易な遺伝子操作導入モデル甲殻類、 Parhyale hawaiensisを示します。
Introduction
fThe CRISPR Cas9 システムに1ゲノムのターゲット領域を変更できます。この迅速かつ安価な技術の基礎研究をもたらしましたし、パーソナライズされた疾患の治療、精密農業、内外2の開発の深遠な影響を作ることを約束します。CRISPR 編集は民主化ツールと、新しい研究室でシステムを実装するゲノム工学、基本的な分子生物学のスキルで特定の専門知識は必要ありません。遺伝子操作3,4のいくつかの代替手段で、以前難治性の生物を研究することができます。5 年だけでは、CRISPR ゲノム編集以上 200 の異なる脊椎動物、無脊椎動物、植物、微生物種をエンジニアに使用されています。
CRISPR 原核生物防御経路から適応部位特異的ゲノムの編集に必要なコア要素s. 化膿から通常の Cas9 タンパク質し、追加の核局在化信号 (NLS) とその特殊なコドン最適化RNA ガイド5,6。ここで説明されていない、他の Cas9 第オーソロガスや CRISPR 酵素も使えます。自然に発生する gRNA は 2 つの個別に転写部分、CRISPR RNA (crRNA) とトランス活性化から成る crRNA (tracrRNA)7。これらの Rna は、シングル ガイド RNA (sgRNA)8として知られている単一のコピーに融合することができます。デュアル ガイドもあるが、ほとんどゲノム編集者選択合理化された sgRNA9,10,11を定期的に使用します。実験者 protospacer 隣接するモチーフ (PAM) と呼ばれる、Cas9 の認識に必要な短いライセンス署名の隣に位置し、確保する 20 ヌクレオチド (nt) ゲノムの DNA のターゲットを選択し、相補的なシーケンス12 を含む gRNA をデザイン.
細胞内 RNP 複合検索そのゲノムのターゲット、gRNA 塩基対の相補的な DNA のストランドし、酵素を切断、両方 dna 二重鎖を生成するは2を破る。細胞修理機械は、少なくとも 2 つのルートのいずれかによって、DSB を修正: エラーが発生しやすい非相同末端結合 (NHEJ) 経路または相同監督修理 (HDR)、経由をシームレスにブレークのいずれかの側に相同性の ' 腕' を含んでいる DNA が組み込まれています。元修理経路塩基形成とそれに伴う遺伝子破壊に後者により実験者を挿入または DNA シーケンス1の変更に通常 します。
編集の効率と精度は、Cas9 と gRNA をセルに入力する手段によって異なります。これらのコンポーネントは、培養細胞、胚、または組立 RNP 複合13,14,15核酸の形で生物に配信場合があります。一般的な核酸ベースの配信方法は、ウイルス伝達、トランスフェクション、または mRNA またはプラスミッド DNA のエレクトロポレーションに含まれます。Cas9 タンパク質と RNA のガイドはセル内で生産し、複合体を形成するのに関連付けられています。
RNP の直接配信 Cas9 蛋白質および RNA のガイドの別の浄化が必要です。これは社内で行うことができます。 または蛋白質と sgRNA は、いくつかの商用ベンダーのいずれかから購入できます。取得されると、Cas9 と gRNA は酵素によって主務 RNP 複合体を形成するために混合し、受精卵/卵、脂質ベースのトランスフェクション16、またはエレクトロポレーションに直接注入によって細胞に導入します。RNP 編集の最初のレポートには、 c. の elegans生殖腺17への注入が関与しています。インジェクションは胚および全有機体の RNP 導入の手段効果的なエレクトロポレーションは、マウス18,19 , ラット20胚で実証されています。我々 は直接注入 RNP線虫 c. エレガンスの生殖腺およびp. hawaiensis胚のプロトコルについて説明してひと初代細胞を編集する際、RNP を配信するエレクトロポレーションの特殊な型をお勧めします。このメソッドは、nucleofection、最適化されたエレクトロポレーション プログラムおよびセル型固有のソリューションが含まれます、RNP 細胞質と核21を入力することができます。
ゲノム編集 RNP でいくつかの明確な利点を提供しています。タンパク質と RNA コンポーネントが完成、納品前に品質を確保することができますので、RNP 編集核酸ベースの配信に関連付けられている多くの落とし穴を回避できます。すなわち、宿主ゲノム DNA の Cas9 エンコード統合のリスクがない、mRNA は劣化にさらされることは、生体内でタンパク質や gRNA 式、折り畳み式、および協会22,23の問題を回避できます。さらに、毒性やプラスミド ベース式、セル24,25,26,27内 RNP の短い半減期の結果よりはるかに少ないオフのターゲット イベントの削減につながる RNP を使用します。
最後に、RNP 編集明白につながるひと細胞、初代培養細胞の様々 な編集率など、線維芽細胞、胚性幹細胞 (Esc)、誘導多能性幹細胞 (iSPCs)、HSPCs、および T 細胞16,24、 25,26,27,28,29;P. hawaiensis、線虫ショウジョウバエ3,17,30を含む無脊椎動物のゼブラフィッシュ、マウス、ラット31,32; のような脊椎動物種で植物のシロイヌナズナ・ タバコ、レタス、米、ブドウ、リンゴ、トウモロコシ ・小麦33,34,35,36; を含む種クラミドモナス、ペニシリウム、カンジダ種37,38,39。RNP プラスミド配信と比較を使用する場合、塩基形成の頻度が高くなることが、HDR を介した DNA の挿入は25,27,29を達成するために簡単にすることができます。
ここで説明したプロトコル Cas9 RNP を使用し、さまざまな生物学的システム40,41, 動作するように困難ではない細胞の特に中に適用は容易容易に適応できる、効果的なテクニックです。正確な遺伝的操作の確立されたシステムを使用しないで有機体と。まず、設計、取得、および別のモデル セル型の有機体の間でその使用をカバーする前に Cas9 RNP をアセンブルする方法について説明します。造血幹細胞/前駆細胞 (HSPCs)、nucleofection、同じメソッドを使用してこのプロトコルの 2 および 3 の手順で一緒に覆われているので T 細胞を編集します。Cの手順を編集します。線虫手順 4 および 5 で説明とP。hawaiensisの編集は、手順 6 と 7 で覆われています。最後に、以来、すべての生物で遺伝子編集の実験の成功は、遺伝子配列による評価が、すべての細胞や生物プロトコルで説明可能な解析方法を説明した手順のステップ 8 の通りです。
Protocol
1. RNP アセンブリ
-
早めに前もってすべての蛋白質、DNA、RNA コンポーネントの取得実験をデザインします。最初のパスとして表 1 肯定的なコントロールの 1 つし、信頼性の高い実験的なデザインと材料の整合性を確保する材料の表に記載されている市販試薬を使用します。新しいゲノム編集実験計画に関する追加ヒントについては、このトピック12,42,43にペーパーを参照してください。
注: 以降の手順で説明するように組み立てられる、結合の事前準備で保存されるかもしれない-80 ° C- ターゲット遺伝子を選択すると、1 つの無料のオンライン ツールを使用して、デザイン、最適な gRNA44,45,の46,47,48。ノックアウトを生成したい場合はエクソンをターゲットしてください。
注: これらのツールは隣接するs. 化膿PAM とターゲット サイトを識別するために役立つシーケンス、高品質スコア、およびオフのターゲット スコアが低い。 - 公開メソッド8までのs. 化膿Cas9 蛋白質を浄化や商用ベンダーから購入。
- RNA の希釈、RNP の準備、KCl、10% グリセロールと TCEP の 1 mM HEPES pH 7.5、150 mM の 20 mM を含むタンパク質ストレージに典型的な Cas9 バッファーを準備します。常に再懸濁しますや劣化を防ぐために RNA を希釈に使用されるバッファーのヌクレアーゼ フリー水を使用します。
- 公開されたメソッドを使用して生体外のトランスクリプションによるガイド RNA (tracrRNA、crRNA または sgRNA) を作ったり、核酸合成会社17,21,49,から購入50,51。
- 遺伝子を挿入するには場合、は、合成またはドナーの DNA のテンプレートを購入します。
- タンパク質と RNA 因数-80 ° C で融解を使用の直前に氷の上に格納します。
注: 各凍結融解は若干効率を低下させます。Cas9 精製52と sgRNAs53の生体外のトランスクリプションのための詳細は、オープン アクセス プロトコルが別の場所であります。
- ターゲット遺伝子を選択すると、1 つの無料のオンライン ツールを使用して、デザイン、最適な gRNA44,45,の46,47,48。ノックアウトを生成したい場合はエクソンをターゲットしてください。
- C. の elegansを操作する場合は、手順 1.5 に進みます。P. hawaiensisプロトコル 1.6 のステップに進みます。SgRNA を使用している場合は、1.4 の手順に進みます。1.3 一次電池を編集するための gRNA を組み立てるためのステップへ進みます。
-
TracrRNA と crRNA の等モル量を混合することによって、gRNA を組み立てます。約 50 のゲノム編集実験のための 80 μ M gRNA 株式の 100 μ L を作る。
- 30 分の 37 ° C で gRNA をインキュベートし、ゆっくりと部屋の温度に冷却します。
-
HSPC と T セルの編集の RNP 準備: 約 30 gRNA (Cas9 バッファーで希釈済み) に集中 Cas9 を追加非常にゆっくりと、10 μ L の総ボリュームの Cas9 蛋白質の 200 pmol に gRNA の 1-2 x のモル量を混合することによって、RNP 複合を組み立てる s、ピペット、20 μ M の最終 Cas9 濃度をもたらす迅速な円を作るします。
- エレクトロポレーション キュベットを準備します。
注: このプロトコルは、材料表に掲げる商業システムに固有、RNP 編集他のエレクトロポレーションのデバイスと達成することができます。 - 各キュベットに 5 μ L (100 pmols, T 細胞) または RNP の 10 μ L (200 pmol、HSPCs) を追加します。
- かどうか、ノックアウトを作るのではなく、新しい DNA を挿入するキュベットやプレートのウェルに 100 μ M (100 pmol) 一本鎖オリゴヌクレオチド ドナー DNA (ssODN)25,,5455の 1 μ L を追加。
- 一次電池の編集では、次の手順の手順 2 にスキッププロトコル。
- エレクトロポレーション キュベットを準備します。
-
RNP線虫を編集するための準備: 20 μ L (最終濃度は、括弧内に記載されています) の最終的なボリュームを作成するために以下の試薬を追加して複雑な要件を組み立てる: Cas9 (2 μ M)、HEPES pH 7.5 (10 μ M)、KCl (115 μ M)、crRNA (12 μ M)、tracrRNA (40 μ M) と、修理のテンプレートに必要な場合(0.5 μ M ssDNA または 350 ng/μ L dsDNA まで)。
注: Cas9 を介した DSB テンプレート修理の効率は dsDNA 修理構成体の濃度に比例したがってより効率的な修理テンプレート濃度が高いテンプレートの修理。ただし、dsDNA の 350 ng/μ L 以上を含む混合物の注入は、注入されたワームの可能性を減らすために示されています。したがって、その致死率を最小限に抑えながら修理効率を最大化するためにミックスに dsDNA の 350 以上 ng/μ L がするように、使用することをお勧めします。- 複数の遺伝子を同時にターゲット、説明する co CRISPR/共同 conversion スクリーニング アプローチに必要な手順で 5.4 に複数の crRNAs を追加します。1 つ以上の crRNA を追加すると、それぞれ順番にマスター ミックスを追加します。
注: 各 crRNA の量は、同じである必要はありません、特定の位置の突然変異の頻度を妨害するも Cas9 の濃度を変更することがなくマスター ミックスで crRNAs の濃度を 2 倍が表示されません。例をペらでくわしく説明します。56。 - ピペッティングで混ぜるし、5 の 16,000 x g で RNP ソリューションをスピン s 管の底でソリューションを収集できることを確認します。
- 15 m の 37 ° C でソリューションを孵化させなさい。
- 薄い退屈マイクロインジェクション針を詰まらせる可能性があります任意の粒子をペレットに 1 分 16,000 x g でサンプルを遠心します。以降の手順で上清を使用します。
- 線虫のプロトコルの残りの部分に対して、手順 4 に進みます。
- 複数の遺伝子を同時にターゲット、説明する co CRISPR/共同 conversion スクリーニング アプローチに必要な手順で 5.4 に複数の crRNAs を追加します。1 つ以上の crRNA を追加すると、それぞれ順番にマスター ミックスを追加します。
-
P. hawaiensis編集の RNP 準備: ヌクレアーゼ フリー水と (注射を可視化) 用フェノールレッド Cas9 と 0.15% フェノールレッドの 6.25 μ M の最終濃度に希釈することで使い捨て Cas9 因数を準備します。
- 6 μ l。 追加 12 pmol Cas9 の gRNA、2 μ M、4-8 μ M の gRNA 濃度 0.05% のフェノールレッド濃度最終 Cas9 濃度をもたらすための総量 Cas9 タンパク質 gRNA の x 2-5 を混合することによって複雑な RNP を組み立てます。
- RNP の複雑に 10 分間、室温で混合物を孵化させなさい。
- P. hawaiensisの編集では、次の手順の手順 6 にスキッププロトコル。
2. 細胞培養そして準備は
注: 生物学的安全キャビネットで 2.1.1 に 3.3.3 の手順に従います。
-
購入凍結人間動員末梢血 CD34+ HSPCs ベンダーから。
- 〜 1 を解凍 x106 HSPCs 37 ° C の水で 3 分のバス ・ 15 mL の円錐管にそれらを転送します。商業ソースから拡張の無血清培地 10 mL を追加し、10 分、上清を除去し、補われた論文の 2 mL の細胞を再懸濁します 100 x g で混合物をスピンします。6 ウェル プレートで細胞をプレートし、RNP エレクトロポレーション前 24-48 h の 37 ° C の定温器でそれらを文化します。
- 診断とセルをカウントし、必要な HSPCs (electroporated にキュベットあたり 150,000-200,000 HSPCs) の合計数を遠心管に転送します。
- 細胞のペレットを 10 分間 100 x g でチューブをスピンします。
-
人間プライマリ CD4 を購入仕入先から+ T 細胞密度勾配遠心法29ひと全血から分離するか。
- T 細胞の活性化の前にあらかじめコート αCD3 48 ウェル文化板 (UCHT1) と αCD28 (CD28.2)。500 μ L の 10 μ G/ml αCD3、37 ° C で少なくとも 2 時間 PBS で 10 μ G/ml αCD28 でプレートをコートします。
注: いくつかの遺伝子座の NHEJ ことがなく実現されます前の刺激が、その効率を最大限にこのステップを含みます。 - ΑCD3/αCD28 抗体結合プレート [RPMI 1640 を添加した 5 mM HEPES の 2 L-グルタミン、商業代わりのペニシリン/ストレプトマイシン、2-メルカプトエタノール、5 mM の 50 μ M の 50 μ g/mL RPMI 完全培地で 37 ° C で 48 時間培養 T 細胞非本質的なアミノ酸、ピルビン酸ナトリウムの 10% (巻/巻) FBS 5 mM]。500 μ L のメディア/48 ウェル プレートのウェルに 2,000,000 の T 細胞の密度で培養 T 細胞。
- 検定と転送を使用してセルを数える T、エレクトロポレーションに必要な T 細胞の総数は、遠心管に (electroporated にキュベットあたり 100,000-1,000,000 T 細胞) を試してみてください。
- 細胞のペレットを 8 分間 90 x g でチューブをスピンします。セルは、密度勾配分離 2 日以内をされている場合は、8 分の 200 x g でスピンします。
- T 細胞の活性化の前にあらかじめコート αCD3 48 ウェル文化板 (UCHT1) と αCD28 (CD28.2)。500 μ L の 10 μ G/ml αCD3、37 ° C で少なくとも 2 時間 PBS で 10 μ G/ml αCD28 でプレートをコートします。
-
両方の細胞の種類の任意の気泡を除去ピペット/真空上清を吸引します。
- 優しくキュベットあたりエレクトロポレーション バッファーの 20 μ L で細胞を再懸濁します。
- すでに泡を作成せず、上下ピペッティングでよく RNP とミックスの 10 μ L を含む各のキュベットに (150,000-200,000 HSPCs または 100,000-1,000,000 T 細胞) の細胞の 20 μ L を追加します。
3. RNP エレクトロポレーション
- Electroporate、nucleofector でそれらを配置した後キュヴェット。HSPCs、パルス コード ER100 を使用します。T 細胞のパルス ・ コード EH 115 を使用します。
-
HSPCs のみ:補完論文媒体 (37 ° C に加温) エレクトロポレーションと 10 ~ 15 分のセルを回復した直後にそれぞれキュベットに追加 100 μ l
- 文化 96 ウェル丸底のプレートし、24 h の補完論文中の追加の 100 μ L を追加するセルを転送します。
- 新鮮な補完論文中にそれらを変更、さらに 24-72 時間インキュベートします。
- 遺伝子型の細胞を除去して 48 96 h のポスト-エレクトロポレーション。5 分間 300 x g でセルをスピンし、DNA の抽出 (8.2 の手順) を開始する前に、上清を除去します。
-
T 細胞のみ: RPMI 80 μ 完了文化メディア マルチ チャンネル ピペットを使用して (必要に応じて) 各キュベットまたは井戸、貯水池からの 37 ° C に加温を追加。
- 37 ° C 15 分でそれらを孵化させなさい。
- 先プレートに適切なメディア、抗体、サイトカイン等を追加し、37 ° C の定温器で暖める前。
- マルチ チャンネル ピペットを使用して (必要に応じて) 丸底 96 ウェル プレートに井戸から 107 μ electroporated セルを転送します。
- 編集の成果を評価する方法については、手順 8 に進みます。
4. c. の elegansの準備
-
マイクロインジェクションの前日: マイクロインジェクションの agarose のパッドを準備します。
- 水にアガロースを追加し、ホット プレートや電子レンジで沸騰させるソリューションをもたらす水の 3% (w/v) アガロース溶液を作る。
- 24 mm × 50 mm × 1.5 mm カバー ガラス スライド テーブルを手配し、ガラス パスツール ピペットを使用してスライドにアガロース溶液の小さな (~ 15 μ L) のドロップを配置します。すぐに agarose ドロップ別 coverslip の上に配置することで平らにします。アガロースを固めるし、coverslips のいずれかを削除するを許可します。
- 卓上乾燥して一晩 agarose コーティング coverslip の顔アップを残します。24 時間後に、清潔で乾燥した容器に agarose パッドを格納します。
注: これらは無期限に使用することができます。
- マイクロインジェクション針を引く: フィラメントとホウケイ酸塩のガラス管を使用して (外径 1.0 mm、内側直径 0.58 mm)、メロ、火災57と58その他のリソースに基づいて針を引きます。針はすぐに使用することができます。 または粘土サポートによるブレース、清潔で乾燥した容器で保存することができます。
- ワームのメンテナンス、ペトリ板に注ぎ、OP50 細菌の斑点のあるマツ材線虫病成長メディア (NGM) 寒天培地を準備 (プロトコルが標準的なc. の elegansメンテナンスと成長媒体のためのレシピは、Stiernagle59を参照してください)。
- マイクロインジェクションのワームをステージ: マイクロインジェクション、前に 12-24 時間 OP50 細菌の新しい NG 寒天プレートに L4 段の両性具有をピックアップし、20 ° C でそれらを一晩インキュベート各 Cas9 ターゲット/注入ミックス プレート 〜 30 ワームを選ぶ。
-
マイクロインジェクションの日:1.5 準備した上澄み RNP ソリューションで引っ張られたマイクロインジェクション針をロードします。
- (一般的に読み込み未満 0.1 μ L) 準備マイクロインジェクション針にキャピラリー ピペットから引っ張られた毛細管ピペットとバックフィル ソリューションへのステップ 1.5.4 から上澄みをピペットします。
- マイクロマニピュレーターに接続されているマイクロインジェクション装置に読み込まれた針をマウントします。250 kPa と 25 kpa バランス圧力噴射装置の圧力を設定します。
-
鋭い針のエッジを生成する読み込まれた針先をブレーク バック。15 mm x 15 mm x 24 mm x 50 mm x 1.5 mm coverslip の上部に 1.5 mm の正方形 coverslip の場所。
- ハロカーボン オイル 700 平方 coverslip の一方の端をオーバーレイします。
- 15 mm の正方形のカバーガラスの端、油に針を置きます。
- 顕微鏡ステージと coverslip ガイド、注入ペダル/ボタンを押したまま、針の端に沿ってスライドをブラシに手を使用してください。針先、針から液体の流れを増やすを破る。〜 1 バブル/s を形成、針の端に沿って流れを混ぜて注射することによって最適な流量を実現します。
- マイクロインジェクションの前に 12-24 時間を選んだ L4 ワーム注入の日に発達段階の若い大人であることを確認します。OP50 細菌を欠いている NG 寒天プレートに若い大人のワームを選ぶし、5 分、周りをクロールできるようにします。これは針下駄を最小限に抑え、注入のパッドに転送される細菌の量を減らします。
- 郭清範囲に agarose インジェクション パッド/coverslip を配置します。ワームのピックを使用してパッドの 1 つの端に沿ってハロカーボン オイルの小さいトラックを置きます。
-
NG 寒天プレートとオイルのトラックにいくつかのワームを持ち上げて油でコーティング ワーム pick を使用する。細い髪、まつげや猫のひげなどのピペットに添付の平行、優しく agarose パッドにワームをプッシュにワームを配置します。までマイクロインジェクション手順に慣れて、マウントのみ、一度に 1 つのワームを注入します。
注: 乾燥アガロースはワーム、パッドに付着させてから水分を芯します。ワームを脱水することができます、したがって、1 つはすぐに働かなければなりません。- 一度の位置およびハロカーボン オイル (~ 20 μ L) ワームの先端から数滴を選ぶ別のワームをオーバーレイ パッドに接続されています。
5 します結合と投与後気にc. の elegans生殖腺マイクロインジェクション。
注: マイクロインジェクション プロトコル Mello および火57から適応され、60,61詳細他の場所で説明されています。
-
インジェクション顕微鏡上にマウントされたワームと coverslip の場所。低倍率 (5 X 目標、10 X 眼)、下で注射針に垂直のワームを配置します。
- 高倍率 (40 × 対物、10 X 眼) への切り替えは、核で半ば-に後半-pachytene 付近に対応する生殖腺の腕に隣接する針を再配置します。
- マニピュレーターを使用すると、キューティクルを少し憂鬱、ワーム針に移動します。その後、片方の手でキューティクルを針の衝撃を顕微鏡ステージ サイドをタップします。インジェクション ペダル/ボタンを押すとゆっくり注入ミックスで生殖腺の腕を埋めるし、針を抜きます。
- その他の生殖腺の腕でこの手順を繰り返します。
-
ワームを注入すると、一度 coverslip/アガロース パッドを取り外して、解剖顕微鏡の下に置きます。
- その上 M9 バッファーをピペッティングによるワームから油を転置する引っ張られた毛細管ピペットを使用して。この処理では、寒天からワームを解放します。
- 10 分後、ワームは、バッファーでどたばたがときに、移動する NG 寒天 OP50 細菌引っ張られた毛細管ピペットを使用して。ワームに入れました、周りに移動するまで 2-3 h の 20 ° C でプレートを配置します。
- 回復、一度個別に OP50 で NG 寒天にワームを転送し、プレートを 25 の ° C の定温器に転送します。
-
P0を許可する-に成長し、3 日間の子孫を置くワームを注入します。画面 F1の子孫。
- Co CRISPR または共同変換62,を使用して,636465,,は検診参照の遺伝子の突然変異体の表現型があるかどうかに基づいて候補ワームを選択します。個別に転送 OP50 で NG 寒天プレートに新しいこれらのマークされたワームと 20 ° C で F2子孫を産むことができます。
注: co CRISPR スクリーニングまたは選択範囲に使用される表現型は、Cas9 編集の成功のため初期見積もりを提供する必要があります。 - Co CRISPR 表現型が存在しない場合は、マイクロインジェクション効率を向上させる支援するために肯定的な制御プラスミドを microinject します。
注: たとえば、エンコード タグ付きの mCherry 墓 2 注入ミックスでプラスミドを含む役立つ注入効率を評価します。正常に pCFJ90 を注入したワームでお越しの際にも蛍光咽頭といくつかの子孫。
- Co CRISPR または共同変換62,を使用して,636465,,は検診参照の遺伝子の突然変異体の表現型があるかどうかに基づいて候補ワームを選択します。個別に転送 OP50 で NG 寒天プレートに新しいこれらのマークされたワームと 20 ° C で F2子孫を産むことができます。
- 必要な編集の有無を F1ワームを確認します。96 ウェル プレートの個々 の井戸に F1母を選ぶ、彼女を溶解し、挿入特定の pcr、dna 塩基配列解析、測量ヌクレアーゼ アッセイ (セル 1)66のいずれかで彼女の DNA を確認します。
注: co CRISPR/共同 conversion または他のスクリーニングまたは選択制度65,66,67,68を使用する場合は、これらの試金を実行できます。 - 編集の成果を評価する方法については、手順 8 に進みます。
6. P. hawaiensis準備
- 前日、マイクロインジェクションを前夜; ペア タンクを設定して初期胚の豊か新しく分離女性新鮮な受精胚が含まれます。Rehmらを参照してください。詳細については69 。
- マイクロインジェクションの日、単一セルParhyale胚を収集 (0-4 h 後受精) 海水中の 0.02% チョウジ油妊娠女性を麻酔して炎プルを使用して彼女の腹の育児嚢から胚を軽くこすると丸みを帯びたガラス ピペットと #3 鉗子の鈍いペア。
7. P. hawaiensis胚マイクロインジェクション結合と投与後のケア
- バックフィル RNP 注入ミックス約 1 μ L で引っ張られたキャピラリー チューブは上記。
-
Rehmらで説明するように、それぞれの胚を microinject に圧縮された窒素を使用します。69。
- マイクロおよびマイクロマニピュレーターを使用して解剖顕微鏡の下でParhyale胚を注入します。引っ張られたキャピラリー チューブ (フィラメント、装置を引っ張ってマイクロ ピペットを使って引くと 4 インチ-1.0 mm) の背面に注入ミックス 1.5 μ を読み込む microloader ピペット チップを使用しています。
- 注射器の針を設定し、切り裂くスコープ下ピンセットのペアを使用して (ごく少量) 針の先端を破る。ハロカーボン油 700 に注入して、気泡の直径を測定によって提供されるボリュームを調整します。
- かみそりの刃を使用して硬化剤のうち「トラフ」をカット。フィルター滅菌海水で途中でそれを埋めるし、安定するトラフParhyale胚をラインアップします。
- マイクロインジェクション セットアップでは、注入時に鉗子のペアでそれぞれの胚を安定化を用いた胚を注入します。注入後、上半フィルター滅菌海水でいっぱい新鮮な 60 mm ディッシュに胚を転送するのに転送する、ガラス製のピペットを使用します。
-
1部フォーム 2 細胞胚 (4 〜 6 時間後受精) に既に発生している場合は、両方の割球を注射することによって完全に突然変異体動物を生成します。2 細胞期の合計胸の谷間 FITC または TRITC デキストランと共同、割球を注入し、信号が注入後スコープを解剖蛍光灯の下で単一割球に制限されていることを確認します。
- また、2 細胞期 (組織および A P 軸に沿った位置によってほぼ分割左-右) で 2 つの割球の 1 つで、注射することによって ' 半分変異体の動物を生成します。
- 単一細菌層に編集の制限 8 細胞胚 (7.5-9 h 後受精) で 1 つのセルを挿入します。チャンレジらを参照してください。初期の割球系統の地図の70 。
-
60 mm 培養皿 (皿あたり 25 以上) をぶるぶると震わせながら、'事前酸素' 水族館バブラーを使用して、フィルター殺菌海水と半いっぱいで胚を孵化させなさい。
- 湿度を維持し、12 時間の明暗サイクルの 26 ° C の定温器の配置に湿紙タオルと並ぶ緩く密封容器に胚の料理を配置します。
- 数日おき海水皿をきれいに存続の胚を転送します。
注: 彼らがはるかに遅く開発は胚が培養、室温で可能性があります。
-
解剖し、その場で交配または抗体染色 (参照・ ブラウンらによって発現解析のさまざまな段階で胚を修正71ステージング ガイドの郭清と固定72の in situハイブリダイゼーション73、74を汚す抗体の追加参照)。
- タングステン線の曲がった部分を約スレッドによって解剖針を作るインスリンの針の端に長さ 0.5。現在の下で水酸化ナトリウムに針を研ぐ。解剖針のハンドルとして 1 mL 注射器を使用します。
- 9 パーツ PEM バッファー (パイプ ph 6.95、グリコールエーテルジアミン四酢酸、2 mM、1 mM MgSO4の 0.1 M)、1 パーツ 10 x PBS および 1部 32% の作りたてのソリューションと 3 もガラスの皿の半分の 1 つの井戸を埋める PFA。3-5 胚を皿に置き、流出する卵黄と定着実行を可能にする、安定させるために突くと少し鈍く鋭いタングステン針を使用して、それぞれの胚に小さな穴を突きます。
- 尖ったタングステン針のペアを使用して、優しくいじめるParhyale胚を取り囲む外側の 2 つの膜。胚をより強固にするための定着剤でそれらを解剖膜除去が難しく、胚に固定になってから膜を保つためにすぐに仕事が。抗体の汚損のための 15-20 分または 40 〜 50 分の in situハイブリダイゼーションのための合計を修正する胚を許可します。
- ライブの孵化の画像し形態および行動の表現型分析または修正しより詳細な分析のため、それらを染色します。ノックアウト、トランスジェニックは行 (雛ケアと他の有用な詳細情報のための Kontarakis と Pavlopoulos75を参照) を確立する 2-3 か月で性成熟に孵化を上げます。
8 評価結果を編集
- 該当する場合は、編集したセルまたは生物の視覚的または機能的な表現型を探します。
メモ: このプロセスは、アプリケーションによって広く異なるが、いくつかの例は、上記に関連するプロトコルの手順の最後で説明します。HSPCs 鎌状赤血球の遺伝子変異を修正した後高速液体クロマトグラフィー (図 1A) を使用して区別された赤芽球によってヘモグロビンの産生を分析します。T 細胞の IL-2 受容体遺伝子のノックアウトは表面の汚損によって確認することができ、フローサイトメトリー (図 1B)。線虫 c. エレガンス P. hawaiensisの表現型を評価するために動物の形態と光や蛍光顕微鏡 (数字 1と1 D) で動作を確認します。 - 効率と生成されたゲノムの編集の種類を決定するには、編集されたセルのプールを溶解し、商業抽出キット21を使用して彼らのゲノム DNA を抽出します。
-
塩基形成の迅速推定、カットの周りを PCR 増幅少なくとも 200 塩基対サイトし、T7 endonuclease1 を実行 (T7E1)76または測量 (CEL 1 ヌクレアーゼ) アッセイ77。
- Cas9 カット サイトまたは成功した HDR で塩基形成が作成または知られている制限サイトを削除、制限の酵素の消化力を使用した編集の効率6の見積もりを検討してください。制限断片長多形 (RFLP) アッセイは、使用する場合、効率を確認する便利な方法をすることができます。
- 編集効率の定量の正確さは、優勢な編集結果の定量 PCR 増幅を送信標準サンガーの前方および逆のプライマー シーケンスします。
注: 単一のクローンまたは有機体の分析、サンガー結果の分析は簡単、図 2Aに示すようです。細胞のプールを分析しは、図 2Bに示すようとオンライン ツール78、クロマト グラムを分析します。 - による定量化と編集結果のシーケンス、図 2Cに示すようにディープ シーケンス27,54, を実行します。
- オフのターゲット変更、PCR 増幅予測オフのターゲット サイトの特定のセットを評価し、NGS の送信。染色体転座の検出を有効にするには、ガイド seq79または高スループット、転流ゲノム シーケンス (HTGTS)80を実行します。クローンの集団でターゲットを編集の全体像、全ゲノム シーケンス (WGS)81,,8283を実行します。
注: は、さまざまな方法がありますの定量化とオフ-ターゲット ゲノム編集、説明さらに様々 なレビュー記事84,85,86の。
Representative Results
これらの実験をどのように組み立て済み Cas9 RNP は初代培養細胞および全有機体のゲノムを操作する使用ことができます。研究者浄化または Cas9 タンパク質と sgRNA を購入、事前、複合体を形成する 2 つのコンポーネントを組み合わせて、セルまたは関心の有機体に、RNP を紹介です。後に発生する (該当する場合) に生まれてくる次の世代の子孫の表現型のチェックや遺伝子型の細胞を収集する編集のための十分な時間を許可します。表現型は、機能アッセイ、式の試金、可視化 (目でまたは顕微鏡)、または実験によって、他の方法を介して観察可能性があります。
鎌状赤血球症を引き起こす β-グロビン変異を修正するために編集されている HSPCs することができます赤血球分化や健康の生産または鎌状ヘモグロビン27,87 (図 1 のなど A)。T 細胞の高親和性 IL-2 受容体遺伝子、CD25 をノックアウトする編集 (IL2RA) の表面染色と流れの cytometry88、分析および機能的 IL-2 刺激 (図 1Bシグナリング応答を検出するために分析できます。).T 細胞は、HIV 感染症89の有効性を含む、異なる表現型の評価が必要多くの臨床的に重要な方法で書き換えることができ、車 T の生体内で抗腫瘍効果細胞11。
線虫みみずは co CRISPR/共同 conversion のスクリーニング方法を使用して、2 つの座位62で同時に編集されています。使う 10参照の遺伝子が、ssODN を使用しての HDR は、テンプレートにより、簡単に得点支配使う 10ゲインの機能突然変異を修復します。ヘテロ F1dpy-10(gof)動物ローラー (Rol)、ホモdpy-10(gof)動物がみすぼらしい (使う)。表現型の存在は、これらの動物の発生編集する Cas9 を示し、Rol または使う F1動物の 2 つ目の軌跡の編集イベントの識別の確率を改善します。編集の実験が成功は Rol または使う9020 以上の F1子孫を降伏注入 P0ワームの 33-50% になります。非 Rol 動物に使う 10を野生型に戻り、関心のホモ編集の選択を選択することが可能ですし。親指のルールとして、co CRISPR 参照の遺伝子をターゲットと crRNA の濃度は、興味の遺伝子をターゲット、crRNA の半分をする必要があります。興味の遺伝子の編集は回復されていない、2 つの CRISPR Rna の比率が望ましい突然変異を回復する可能性を高めるに調整できます。たとえば、参照の遺伝子の位置で編集をまた所有しているワームの人口内の興味の遺伝子の編集を持つワームの割合増える参照遺伝子 crRNA と関連する関心の遺伝子の crRNA の量を増やします。共同変換周波数は異なりますが、料金は、通常、20 〜 60%、しばしば F1世代 (図 1C) ホモ編集を降伏します。
P. hawaiensisの孵化 (Abd B)腹部 B遺伝子をノックアウトする編集されているが明確な形態学的異常3 (図 1D) を表示します。この遺伝子は正しい腹部パターニングのため必要で、腹部に存在する呼吸器型ジャンプと歩行脚は、通常、スイミングとアンカーの脚を交換にその中断の結果。
ゲノム遺伝子型のレベルで結果を編集を決定するには、シーケンスまたはシーケンスの変更を検出するの in vitroアッセイが必要です。ここでは、私たちは私たちのモデル細胞の種類や生物から数量を編集する別のアプローチを強調表示代表的なシーケンス データを表示します。ここに示すすべてのメソッドは、任意の生物学的システムに適用できるため図ラベルが一般化されることに注意してください。
シーケンス ベースのアプローチは、技術的な複雑さおよび結果の深さで変わる。クローンの編集集団、容易に分離できる個々 の生物のゲノム DNA 抽出編集した個人を配置できます。Cas9 カット地点とその機能 (図 2A) を混乱させるだろう仮説 frameshifts 与えられた個々 のシーケンス変更標準サンガーのシーケンスの結果を明らかにします。シーケンス処理に使用するオンライン ツール別サンガー法を用いたアプローチです個々 の変異体78よりもむしろ混合集団に適用することができます。シーケンスは、全体的な編集の効率として優勢なシーケンス結果に近づけることができるオンラインのツールと分析されます。代表的なデータは、図 2Bのとおりです。
ここで説明する最も完全なシーケンス方法はディープ シーケンス (高スループットまたは次世代シーケンス処理とも呼ばれます)。このメソッドは、混合された人口の個々 のゲノムから DNA シーケンスを提供します。このようなデータは、さまざまな方法で示すことができます。ここでは、我々 は編集の結果 (図 2C) に基づいて編集されたセルから個別シーケンスの読み込みを分類しています。ほとんどの細胞は、通常遺伝子破壊の結果 NHEJ 経路を介して編集されています。他では、ターゲット遺伝子スワップ アウトされている HDR27を介して別のバージョンの。
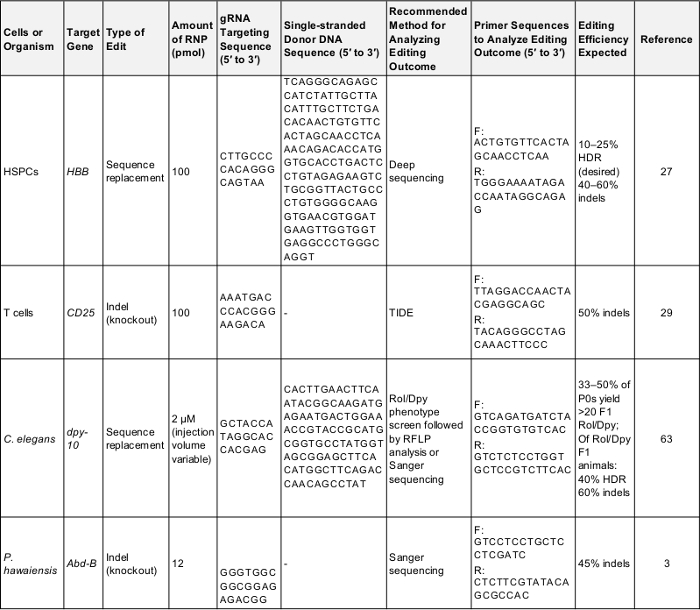
表 1: 陽性コントロール予備ゲノム実験を編集します。このテーブルは、それぞれの細胞や生物はこのプロトコルで記述されている実験の編集初めてゲノムを実行に必要な重要な情報を示しています。プロトコルをテストするために使用できる正常な結果を得られない可能性がこれらのパラメーター、次または一度に比較用のベースラインとして実験者が、自分の興味の遺伝子を狙っています。F: を転送、r: 逆に、HDR: 相同性監督の修理。この表をダウンロードするにはここをクリックしてください。

図 1: Cas9 RNP 編集のひと初代細胞や生物に起因する表現型の代表。(A)これは成功したゲノム編集、後期赤芽球に区別される HSPCs 鎌状ヘモグロビンよりもより機能的なヘモグロビンが生成されます後ことを示す高速液体クロマトグラフィー トレース。正常に編集の細胞は胎児のヘモグロビン (HbF) だけでなく、健全なヘモグロビン (HbA、HbA2) を生産している、突然変異体の赤血球は鎌状ヘモグロビン (HbS) を生成します。任意の単位 (au) で吸光度のグラフを示します。デウィット et らに発表されたこのパネル27. それは科学の進歩のためのアメリカの協会から許可を得て転載。(B)条件ごとに、左側のこのパネルはCD25遺伝子が RNP でノックアウトされた後、T 細胞の表面染色で CD25 を表現しないことを示す流れ cytometry データを示しています。CD25 豊富は、y 軸上のセル サイズを x 軸にプロットされます。条件ごとに、右側は、このパネルは、IL-2 に誘導後リン Stat5 (pStat5) 数量を示しています。シグナリングは、IL-2 受容体 (CD25 KO) 欠席が削減されます。PStat5 豊かさは x 軸にプロットされ、IL-2 入力の 3 つのレベルから生じるデータが垂直方向に比較されます。(C)このパネル画面を示す線虫co CRISPR/共同 conversion 共同コンバージョン マーカーとして使う 10のターゲットします。2 ガイド Rna ターゲット 2 座、使う 10あなたのお気に入りの遺伝子 (yfg) は同じ P0-挿入された動物。使う 10の HDR 結果 Rol または使う表現です。Rol-使う F1動物の選択で 2 つ目の軌跡の編集を識別する機会が増えます。(D)このパネルは、野生型Parhyale hawaiensis孵化がある通常腹部水泳やアンカーの脚を示しています。アブド Bノックアウト孵化 (F0個人) 開発腹部胸部へ変換します。したがって、スイミングとアンカーの脚では、なくなっているし、通常胸部に関連付けられているジャンプおよび歩行の足に置き換えられます。この図の拡大版を表示するのにはここをクリックしてください。

図 2: 結果分析メソッドの編集からの典型的な結果。(A)このパネルは、野生型のシーケンスと開いたリーディング ・ フレームをシフトすることによって遺伝子の機能を混乱させる 3 つの異なるオクターリピートなどを含む個々 の F1 P. hawaiensis生物から結果を配列、サンガーの例を示します。(B)これら潮結果の挿入およびシーケンスされた T 細胞のプールに Cas9 ターゲット ・ サイトで発生した削除イベントの範囲を示します。X 軸は、ヌクレオチドの特定の挿入削除の長さを示します。(C)これらのディープ シーケンスの結果を示さないゲノム編集 nucleofection または gRNA、なし、成功したそのまま Cas9 RNP で編集、HSPCs の DNA 修復結果でグループ化この図の拡大版を表示するのにはここをクリックしてください。
Discussion
堅牢なゲノム編集プロトコルを確立するセルの行または興味の生物で、最適化が必要し、経験的ないくつかの重要なパラメーターのテスト、このセクションで説明します。ここで紹介する一般的な方法のいくつかのバリエーションをしようとして推奨されています。このプロトコルの重要な制限は、これらのメソッドを他のセルに適用することや生物の研究、種によって異なる結果につながる可能性があります高効率遺伝子ノックアウトにつながる実験デザインは DNA の挿入を働きかけることはできません。したがって、ここで紹介され以下のようにトラブルシューティング メソッドで始まるをお勧めします。
ゲノム編集試薬品質のトラブルシューティング。
生成や高品質の試薬を購入は、プロトコルを編集すべてのゲノムの重要なステップです。Cas9 タンパク質をラボで精製または商業的に購入できます。多くのプロトコルが RNP のレシピで、Cas9 の最終濃度を注意してください、最適な遺伝子の活動を編集はソースによって異なりますが、個々 Cas9 タンパク質製剤の特定のアクティビティに依存します。ここで提示されたプロトコルを使用すると、一度検討 RNP Cas9 レベルの滴定によって最適な濃度を確立するために使用量を最適化する: 1 つによって引き起こされる不必要なターゲットを胸の谷間に非常に特定のターゲット DNA 切断を提供します。過剰な Cas940。
ガイド RNA の純度と均質性もゲノム編集成功22の決定要因をすることができます。購入した sgRNAs または crRNA と tracrRNA の個別のコンポーネントは、一般的に高品位の試薬、さまざまな化学修飾は RNA の劣化の問題を戦うためや RNP91に追加機能を吹き込むため可能です。化学修飾した gRNAs は、標準的なゲノム実験を編集に必要なことができない、いくつかのグループがはるかに高い場合や、彼らはプロセスをマスターした後に試して価値がある可能性がありますので、このような試薬により、効率性を編集観察 gRNA 劣化問題22,91に表示されます。In vitro転写と後続のゲルの浄化は、安価な代わりに、ルーチンのゲノム実験17,21,49,50を編集のために十分となります。さらに、同種の gRNA 集団体内など個々 のガイドのリボザイムと tRNA ベース切除を生成する一般的に適用される、体外にクリーナーを生成する RNA の準備を拡張する可能性がありますいくつかのアプローチ製品92。
ガイド RNA と DNA のドナーのデザインのヒント。
ガイド RNA 選択は、非常に効率的なターゲットでは、ターゲットを胸の谷間の可能性を最小限に抑えながらの編集の達成の重要な要因です。ガイドの選択を支援するためいくつかの研究は成功ガイド47,79,93,94、シーケンス機能をコンパイルするのに次世代シーケンサーと相まって高速画面を使用しています。 95,96。これらの機能は、予測アルゴリズムとガイド選択44,45,46,47,48を支援するオンライン ツールを開発する使用されています。このようなアルゴリズムは、ガイド RNA 発現の DNA ベースのシステムを使用して画面に接地されます。Pol III プロモーターを使用してガイドを表現し、彼らの表現が Pol III 転写、早期終了のウラシル97,98、トラックが発生した場合などに関連付けられている制限になりやすいため 99。ただし、結合の使用は体外で作られて-合成ガイド Rna これらの懸念をバイパスし、簡単にガイド設計上の制約。これらのアルゴリズムから登場し、非常に効果的なゲノムの編集、多数の調査で確認されている一般的な機能は、プリン、特にのグアニンは、ガイドのターゲット特定シーケンスの 3 ' 端の存在です。このガイドの機能は、哺乳類から線虫ショウジョウバエ、ゼブラフィッシュ65,100,101まで及ぶ有機体の間で非常に成功しています。また、線虫ガイドのターゲット領域の 3 ' 端に GG 型とガイドの設計は、非常に効果的なガイド Rna65を予測するための効果的な戦略。理想的は特定のアプリケーションに最も成功したを判断する並列で複数のガイドをテストします。
DNA 配列をゲノムに導入しようとすると、ドナーまたはテンプレート DNA の設計は重要なまたです。他の典型的な修理のテンプレート、線形二重鎖とプラスミド DNA54,55,102よりも確実に一本鎖オリゴヌクレオチド ドナー (ssODNs) が挿入されます。いくつかの遺伝子座で、非標的を補足 DNA の繊維を転置または長さ27,55で非対称である相同性腕を所有するいると ssODNs と HDR の効率を改善できます。修理テンプレートですがカットのサイトに挿入される目標とされたシーケンスが含まれていますので、手順は、Cas9 のゲノムの挿入前後にドナー DNA を切断を防ぐために取られなければなりません。これは挿入された遺伝子21,103の機能を維持しながら Cas9 による認識を回避する PAM シーケンスまたは種子地域にサイレント突然変異を作るします。PAM にも単一のヌクレオチドの変更がバインディング104を廃止する可能性がありますが、念のために少なくとも 4 つのヌクレオチドを変更しようとするとします。
意義と将来のアプリケーション:
ゲノム編集 CRISPR Cas9 は、すべての生物の安易な遺伝子操作を有効にする強力な方法として浮上しています。Cas9 RNP で編集最初に少し手間がかかりますが、試薬、プロトコルはラボで確立されるを使用する簡単です。高い全体的な編集の効率、少ないオフターゲット効果24,25,26 HDR、を介して達成困難な遺伝子挿入を含むにつながるプラスミド DNA ではなく組み立て済み RNP でセルの編集,27,29します。 さらに、実験者は遺伝子発現、RNA 分解、タンパク質の折り畳み、および gRNA と別々 に合成されるセル22,23Cas9 分子間の関連付けの問題を回避します。また挿入突然変異誘発について安全上の懸念を回避 RNP 編集して臨床的に14] ウイルスの配信方法は、するときに発生する持続的な表現を使用します。これらの利点のため多くの科学者が前臨床試験の実施概念実証実験を支持する RNP のひと治療用編集します。体内と体外の両方の要件に基づくゲノム編集アプローチは、治療もさまざまな条件、デュシェンヌ型筋ジストロフィー105と鎌状赤血球症、27のような遺伝的疾患を開発HIV の29と癌の11。興味深いことに、Cas9 RNP はますます植物33,34,36の編集 'DNA 自由' を有効にするために、農業工学のための配信方法として採用されています。
Disclosures
著者アレクサンダー マルソンとジェイコブ E. トウモロコシは、スポット ライト治療の共同創設者です。ジェイコブ E. トウモロコシはミッション治療アドバイザーと彼の研究室は、アストラゼネカ、ファイザー社から受託研究の支援を受けた。アレクサンダー マルソンはジュノ治療学協定、顧問と彼の研究室は、ジュノ治療、Epinomics、およびサノフィから受託研究の支援を受けた。彼の研究室は、Cas9 RNP 技術に関連する特許に申請しました。
Acknowledgments
私たちのラボとこれらの方法の開発への貢献のためのベイエリア ゲノム編集コミュニティの多くの前のメンバーに感謝いたします。批判的にこの原稿を読んで、ロス ・ ウィルソンに感謝します
アレクサンドル ・ マルソンの研究はジェイク Aronov からの贈り物によってサポートされ、国民の多数硬化の社会 (CA 1074-A-21) を付与します。アレクサンダー マルソン バローズ Wellcome の基金から医療科学者のキャリア賞を保持して、チャン Zuckerberg Biohub 捜査官です。ジェイコブ E. トウモロコシの研究は、再生医学の李の Ka Shing 財団、医学研究の遺産医療研究所、カリフォルニア工科大学によってサポートされます。Behnom Farboud ・ バーバラ ・ j ・ マイヤーの研究はハワード ヒューズの医学の協会の調査官であるバーバラ ・ j ・ マイヤーに日の出付与 R01 GM030702 によって一部で賄われて。エリン ・ ジャービスと Nipam h. パテルの研究は NSF grant IOS 1257379 によって一部で賄われてやエリン ・ ジャービスは、NSF GRFP と Philomathia 大学院フェローシップからサポートを認めています。
Materials
| Name | Company | Catalog Number | Comments |
| Reagents/Materials | |||
| DNA oligonucleotides | Integrated DNA Technologies | - | IDT will provide custom DNA sequences, including those in Table 1 |
| Guide RNAs | Synthego | - | Synthego will provide high-quality sgRNAs for S. pyogenes Cas9, including custom sgRNAs containing the targeting sequences included in Table 1 |
| Purified Cas9 protein (EnGen Cas9 NLS, S. pyogenes) | New England Biosciences | M0646T | If possible, purifying Cas9 in-house or purchasing from local core facilities is a less expensive option |
| Normal peripheral blood CD34+ stem/progenitor cells | AllCells | PB032-2 | |
| StemSpan SFEM | StemCell Technologies | 09650 | |
| StemSpan CC110 | StemCell Technologies | 02697 | |
| P3 Primary Cell 4D-Nucleofector X Kit | Lonza | V4XP-3032 | |
| RPMI-1640 Medium, With sodium bicarbonate, without L-glutamine, liquid | Sigma | R0883-6X500ML | |
| EasySep™ Human T Cell Isolation Kit | Stemcell | 17951 | |
| cell culture plate, 96 wells, round | Fisher Scientific | 3799 | |
| CTS (Cell Therapy Systems) Dynabeads CD3/CD28 | Life Tech | 40203D | |
| Reombinant Human IL-2 | UCSF Pharmacy | NA | |
| SepMate-50 500-pack IVD | Stemcell Technologies | 85460 | |
| OP50 Escherichia coli | Caenorhabditis Genetics Center | OP-50 | https://cgc.umn.edu/ |
| Nematode Growth Media agar in petri dishes | - | - | See Stiernagle, T (ref. 59) |
| Standard borosilicate glass capillaries with filament: 4 in (100 mm), 1/0.58 OD/ID | World Precision Instruments | 1B100F-4 | |
| Single-barrel standard borosilicate glass capillaries: 6 in (152 mm), 2/1.12 OD/ID | World Precision Instruments | 1B200-6 | |
| Cover glass; 24 × 50 mm | Thermo Fisher Scientific | 12-544E | |
| Cover glass; 22 × 22 mm | Thermo Fisher Scientific | 12-518-105K | |
| Apex LE agarose | Genesee Scientific | 20-102 | |
| Halocarbon oil 700 | Sigma-Aldrich | H8898-100ML | |
| pCFJ90 plasmid | Addgene | 19327 | |
| Compressed nitrogen | - | ||
| 60 mM culture dishes | BD | ||
| Capillary tubes with filament: 4 in (1.0 mm) | World Precision Instruments | T2100F-4 | |
| Sylgard 184 | Dow Corning | ||
| Petri dishes (100 × 15 mm) | - | ||
| Tungsten wire (0.005 in. diameter) | Ted Pella | ||
| Perfluoroalkoxy alkane (PFA) | - | ||
| Marine salt | - | ||
| 9" pasteur pipettes | - | ||
| Phenol red | - | ||
| Nuclease-free water | - | ||
| Equipment | |||
| 4D Nucleofector | Lonza | AAF-1002X | |
| MZ75 Stereomicroscope | Leica | Out-of-production. Current model is the M80 Stereomicroscope | |
| Axio Vert35 inverted phase contrast fluorescent microscope | Zeiss | Out-of-production. Current model is the Axio VertA.1 | |
| Laser-based micropipette puller (for C. elegans protocol) | Sutter Instrument | FG-P2000 | |
| Picoliter Microinjector (for C. elegans protocol) | Warner Instruments | PLI-100A | |
| Three-axis Joystick oil hydraulic micromanipulator | Narishige International | MO-202U | |
| Coarse manipulator | Narishige International | MMN-1 | |
| Micropipette puller (for P. hawaiensis protocol) | Sutter Instrument | P-80/PC | |
| Microinjector (for P. hawaiensis protocol) | Narishige | IM300 | |
| Microloader pipette tips | Eppendorf | 5242956003 | |
| NG-agar |
References
- Komor, A. C., Badran, A. H., Liu, D. R. CRISPR-based technologies for the manipulation of eukaryotic genomes. Cell. , 1-17 (2016).
- Barrangou, R., Horvath, P. A decade of discovery: CRISPR functions and applications. Nature Microbiology. 2, 1-9 (2017).
- Martin, A., Serano, J. M., et al. CRISPR/Cas9 mutagenesis reveals versatile roles of Hox genes in crustacean limb specification and evolution. Current Biology. 26 (1), 14-26 (2016).
- Goldstein, B., King, N. The future of cell biology: emerging model organisms. Trends in Cell Biology. 26 (11), 818-824 (2016).
- Mali, P., Yang, L., et al. RNA-guided human genome engineering via Cas9. Science. 339 (6121), 823-826 (2013).
- Cong, L., Ran, F. A., et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 339 (6121), 819-823 (2013).
- Deltcheva, E., Chylinski, K., et al. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature. 471 (7340), 602-607 (2011).
- Jinek, M., Chylinski, K., Fonfara, I., Hauer, M., Doudna, J. A., Charpentier, E. A Programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 337 (6096), 816-821 (2012).
- Nowak, C. M., Lawson, S., Zerez, M., Bleris, L. Guide RNA engineering for versatile Cas9 functionality. Nucleic Acids Research. 44 (20), 9555-9564 (2016).
- Jiang, W., Cox, D., Zhang, F., Bikard, D., Marraffini, L. A. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nature Biotechnology. , 1-9 (2013).
- Rupp, L. J., Schumann, K., et al. CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Scientific Reports. 7 (1), 737 (2017).
- Graham, D. B., Root, D. E. Resources for the design of CRISPR gene editing experiments. Genome Biology. 16, 260 (2015).
- Wang, H., La Russa, M., Qi, L. S. CRISPR/Cas9 in genome editing and beyond. Annual Review of Biochemistry. 85, 2270 (2016).
- Nelson, C. E., Gersbach, C. A. Engineering delivery vehicles for genome editing. Annual Review of Chemical and Biomolecular Engineering. 7, 637-662 (2016).
- Yin, H., Kauffman, K. J., Anderson, D. G. Delivery technologies for genome editing. Nature Reviews Drug Discovery. 16 (6), 387-399 (2017).
- Zuris, J. A., Thompson, D. B., et al. Cationic lipid-mediated delivery of proteins enables efficient protein-based genome editing in vitro and in vivo. Nature Biotechnology. 33 (1), 73-80 (2015).
- Cho, S. W., Lee, J., Carroll, D., Kim, J. -S., Lee, J. Heritable gene knockout in Caenorhabditis elegans by direct injection of Cas9-sgRNA ribonucleoproteins. Genetics. 195 (3), 1177-1180 (2013).
- Wang, W., Kutny, P. M., et al. Delivery of Cas9 protein into mouse zygotes through a series of electroporation dramatically increases the efficiency of model creation. Journal of Genetics and Genomics. 43 (5), 319-327 (2016).
- Chen, S., Lee, B., Lee, A. Y. -F., Modzelewski, A. J., He, L. Highly efficient mouse genome editing by CRISPR ribonucleoprotein electroporation of zygotes. Journal of Biological Chemistry. 291 (28), 14457-14467 (2016).
- Remy, S., Chenouard, V., et al. Generation of gene-edited rats by delivery of CRISPR/Cas9 protein and donor DNA into intact zygotes using electroporation. Scientific Reports. 7 (1), 16554 (2017).
- DeWitt, M. A., Corn, J. E., Carroll, D. Genome editing via delivery of Cas9 ribonucleoprotein. Methods. , 1-7 (2017).
- Hendel, A., Bak, R. O., et al. Chemically modified guide RNAs enhance CRISPR-Cas genome editing in human primary cells. Nature Biotechnology. 33 (9), 985-989 (2015).
- Thyme, S. B., Akhmetova, L., Montague, T. G., Valen, E., Schier, A. F. Internal guide RNA interactions interfere with Cas9-mediated cleavage. Nature Communications. 7, 11750 (2016).
- Kim, S., Kim, D., Cho, S. W., Kim, J., Kim, J. -S. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Research. 24 (6), 1012-1019 (2014).
- Lin, S., Staahl, B. T., Alla, R. K., Doudna, J. A. Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. eLife. 3, 04766 (2014).
- Liang, X., Potter, J., et al. Rapid and highly efficient mammalian cell engineering via Cas9 protein transfection. Journal of Biotechnology. 208, 44-53 (2015).
- DeWitt, M. A., Magis, W., Bray, N. L., Wang, T. Selection-free genome editing of the sickle mutation in human adult hematopoietic stem/progenitor cells. Science Translational Medicine. 8 (360), (2016).
- Ramakrishna, S., Kwaku Dad, A. -B., Beloor, J., Gopalappa, R., Lee, S. -K., Kim, H. Gene disruption by cell-penetrating peptide-mediated delivery of Cas9 protein and guide RNA. Genome Research. 24 (6), 1020-1027 (2014).
- Schumann, K., Lin, S., et al. Generation of knock-in primary human T cells using Cas9 ribonucleoproteins. Proceedings of the National Academy of Sciences of the United States of America. 112 (33), 10437-10442 (2015).
- Lee, J. -S., Kwak, S. -J., et al. RNA-guided genome editing in Drosophila with the purified Cas9 protein. G3: Genes, Genomes, Genetics (Bethesda, MD). 4 (7), 1291-1295 (2014).
- Sung, Y. H., Kim, J. M., et al. Highly efficient gene knockout in mice and zebrafish with RNA-guided endonucleases. Genome Research. 24 (1), 125-131 (2014).
- Menoret, S., De Cian, A., et al. Homology-directed repair in rodent zygotes using Cas9 and TALEN engineered proteins. Scientific Reports. 5, 14410 (2015).
- Woo, J. W., Kim, J., et al. DNA-free genome editing in plants with preassembled CRISPR-Cas9 ribonucleoproteins. Nature Biotechnology. 33 (11), 1162-1164 (2015).
- Malnoy, M., Viola, R., et al. DNA-free genetically edited grapevine and apple protoplast using CRISPR/Cas9 ribonucleoproteins. Frontiers in Plant Science. 7, 1904 (2016).
- Svitashev, S., Schwartz, C., Lenderts, B., Young, J. K., Mark Cigan, A. Genome editing in maize directed by CRISPR-Cas9 ribonucleoprotein complexes. Nature Communications. 7, 13274 (2016).
- Liang, Z., Chen, K., et al. Efficient DNA-free genome editing of bread wheat using CRISPR/Cas9 ribonucleoprotein complexes. Nature Communications. 8, 14261 (2017).
- Shin, S. -E., Lim, J. -M., et al. CRISPR/Cas9-induced knockout and knock-in mutations in Chlamydomonas reinhardtii. Scientific Reports. 6, 27810 (2016).
- Pohl, C., Kiel, J. A. K. W., Driessen, A. J. M., Bovenberg, R. A. L., Nygård, Y. CRISPR/Cas9 based genome editing of Penicillium chrysogenum. ACS Synthetic Biology. 5 (7), 754-764 (2016).
- Grahl, N., Demers, E. G., Crocker, A. W., Hogan, D. A. Use of RNA-protein complexes for genome editing in non-albicans Candida species. mSphere. 2 (3), (2017).
- Rivera-Torres, N., Kmiec, E. B. A standard methodology to examine on-site mutagenicity as a function of point mutation repair catalyzed by CRISPR/Cas9 and ssODN in human cells. Journal of Visualized Experiments. (126), (2017).
- Nandal, A., Mallon, B., Telugu, B. P. Efficient generation and editing of feeder-free IPSCs from human pancreatic cells using the CRISPR-Cas9 system. Journal of Visualized Experiments. (129), (2017).
- Mohr, S. E., Hu, Y., Ewen-Campen, B., Housden, B. E., Viswanatha, R., Perrimon, N. CRISPR guide RNA design for research applications. The FEBS Journal. 283 (17), 3232-3238 (2016).
- Bauer, D. E., Canver, M. C., Orkin, S. H. Generation of genomic deletions in mammalian cell lines via CRISPR/Cas9. Journal of Visualized Experiments. (95), e52118 (2015).
- Hsu, P. D., Scott, D. A., et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nature Biotechnology. 31 (9), 827-832 (2013).
- Heigwer, F., Kerr, G., Boutros, M. E-CRISP: fast CRISPR target site identification. Nature Methods. 11 (2), 122-123 (2014).
- Moreno-Mateos, M. A., Vejnar, C. E., et al. CRISPRscan: designing highly efficient sgRNAs for CRISPR-Cas9 targeting in vivo. Nature Methods. 12 (10), 982-988 (2015).
- Labun, K., Montague, T. G., Gagnon, J. A., Thyme, S. B., Valen, E. CHOPCHOP v2: a web tool for the next generation of CRISPR genome engineering. Nucleic Acids Research. 44, 272-276 (2016).
- Haeussler, M., Schönig, K., et al. Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biology. 17 (1), 148 (2016).
- Lo, T. -W., Pickle, C. S., et al. Precise and heritable genome editing in evolutionarily diverse nematodes using TALENs and CRISPR/Cas9 to engineer insertions and deletions. Genetics. 195 (2), 331-348 (2013).
- Bassett, A., Liu, J. -L. CRISPR/Cas9 mediated genome engineering in Drosophila. Methods. 69 (2), 128-136 (2014).
- Prior, H., Jawad, A. K., MacConnachie, L., Beg, A. A. Highly efficient, rapid and co-CRISPR independent genome editing in Caenorhabditis elegans. G3: Genes, Genomes, Genetics. , Bethesda, MD. (2017).
- Hirsh, A. Cas9 expression and purification protocol. protocols.io. , (2017).
- DeWitt, M. A., Wong, J. In vitro transcription of guide RNAs. protocols.io. , (2017).
- Yang, L., Guell, M., et al. Optimization of scarless human stem cell genome editing. Nucleic Acids Research. 41 (19), 9049-9061 (2013).
- Richardson, C. D., Ray, G. J., DeWitt, M. A., Curie, G. L., Corn, J. E. Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA. Nature Biotechnology. 34 (3), 339-344 (2016).
- Paix, A., Folkmann, A., Seydoux, G. Precision genome editing using CRISPR-Cas9 and linear repair templates in C. elegans. Methods. 121-122, 86-93 (2017).
- Mello, C., Fire, A. DNA transformation. Methods in Cell Biology. 48, 451-482 (1995).
- Sutter Pipette Cookbook. , Available from: https://www.sutter.com/PDFs/pipette_cookbook.pdf (2017).
- Stiernagle, T. Maintenance of C. elegans. WormBook: the online review of C. elegans biology. , (2006).
- Evans, T. C. Transformation and microinjection. WormBook: the online review of C. elegans biology. , (2006).
- Berkowitz, L. A., Knight, A. L., Caldwell, G. A., Caldwell, K. A. Generation of stable transgenic C. elegans using microinjection. Journal of Visualized Experiments. (18), (2008).
- Kim, H., Ishidate, T., et al. A co-CRISPR strategy for efficient genome editing in Caenorhabditis elegans. Genetics. 197 (4), 1069-1080 (2014).
- Arribere, J. A., Bell, R. T., Fu, B. X. H., Artiles, K. L., Hartman, P. S., Fire, A. Z. Efficient marker-free recovery of custom genetic modifications with CRISPR/Cas9 in Caenorhabditis elegans. Genetics. 198 (3), 837-846 (2014).
- Ward, J. D. Rapid and precise engineering of the Caenorhabditis elegans genome with lethal mutation co-conversion and inactivation of NHEJ repair. Genetics. 199 (2), 363-377 (2015).
- Farboud, B., Meyer, B. J. Dramatic enhancement of genome editing by CRISPR/Cas9 through improved guide RNA design. Genetics. 199 (4), 959-971 (2015).
- Wood, A. J., Lo, T. -W., et al. Targeted genome editing across species using ZFNs and TALENs. Science. 333 (6040), 307 (2011).
- Friedland, A. E., Tzur, Y. B., Esvelt, K. M., Colaiácovo, M. P., Church, G. M., Calarco, J. A. Heritable genome editing in C. elegans via a CRISPR-Cas9 system. Nature Methods. 10 (8), 741-743 (2013).
- Dickinson, D. J., Ward, J. D., Reiner, D. J., Goldstein, B. Engineering the Caenorhabditis elegans genome using Cas9-triggered homologous recombination. Nature Methods. 10 (10), 1028-1034 (2013).
- Rehm, E. J., Hannibal, R. L., Chaw, R. C., Vargas-Vila, M. A., Patel, N. H. Injection of Parhyale hawaiensis blastomeres with fluorescently labeled tracers. Cold Spring Harbor Protocols. 2009 (1), (2009).
- Gerberding, M., Browne, W. E., Patel, N. H. Cell lineage analysis of the amphipod crustacean Parhyale hawaiensis reveals an early restriction of cell fates. Development (Cambridge, England). 129 (24), 5789-5801 (2002).
- Browne, W. E., Price, A. L., Gerberding, M., Patel, N. H. Stages of embryonic development in the amphipod crustacean, Parhyale hawaiensis. Genesis. 42 (3), New York, NY. 124-149 (2005).
- Rehm, E. J., Hannibal, R. L., Chaw, R. C., Vargas-Vila, M. A., Patel, N. H. Fixation and dissection of Parhyale hawaiensis embryos. Cold Spring Harbor Protocols. 2009 (1), (2009).
- Rehm, E. J., Hannibal, R. L., Chaw, R. C., Vargas-Vila, M. A., Patel, N. H. In situ hybridization of labeled RNA probes to fixed Parhyale hawaiensis embryos. Cold Spring Harbor Protocols. 2009 (1), (2009).
- Rehm, E. J., Hannibal, R. L., Chaw, R. C., Vargas-Vila, M. A., Patel, N. H. Antibody staining of Parhyale hawaiensis embryos. Cold Spring Harbor Protocols. 2009 (1), (2009).
- Kontarakis, Z., Pavlopoulos, A. Transgenesis in non-model organisms: the case of Parhyale. Methods in Molecular Biology. 1196, Clifton, NJ. 145-181 (2014).
- Kim, H. J., Lee, H. J., Kim, H., Cho, S. W., Kim, J. -S. Targeted genome editing in human cells with zinc finger nucleases constructed via modular assembly. Genome Research. 19 (7), 1279-1288 (2009).
- Qiu, P., Shandilya, H., D'Alessio, J. M., O'Connor, K., Durocher, J., Gerard, G. F. Mutation detection using Surveyor nuclease. BioTechniques. 36 (4), 702-707 (2004).
- Brinkman, E. K., Chen, T., Amendola, M., van Steensel, B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Research. 42 (22), 168 (2014).
- Tsai, S. Q., Zheng, Z., et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nature Biotechnology. 33 (2), 187-197 (2015).
- Frock, R. L., Hu, J., Meyers, R. M., Ho, Y. -J., Kii, E., Alt, F. W. Genome-wide detection of DNA double-stranded breaks induced by engineered nucleases. Nature Biotechnology. 33 (2), 179-186 (2015).
- Smith, C., Gore, A., et al. Whole-genome sequencing analysis reveals high specificity of CRISPR/Cas9 and TALEN-based genome editing in human iPSCs. Cell Stem Cell. 15 (1), 12-13 (2014).
- Veres, A., Gosis, B. S., et al. Low incidence of off-target mutations in individual CRISPR-Cas9 and TALEN targeted human stem cell clones detected by whole-genome sequencing. Cell Stem Cell. 15 (1), 27-30 (2014).
- Kim, D., Bae, S., et al. Digenome-seq: genome-wide profiling of CRISPR-Cas9 off-target effects in human cells. Nature Methods. 12 (3), 237-243 (2015).
- Hendel, A., Fine, E. J., Bao, G., Porteus, M. H. Quantifying on- and off-target genome editing. Trends in Biotechnology. 33 (2), 132-140 (2015).
- O'Geen, H., Yu, A. S., Segal, D. J. How specific is CRISPR/Cas9 really. Current Opinion in Chemical Biology. 29, 72-78 (2015).
- Tsai, S. Q., Joung, J. K. Defining and improving the genome-wide specificities of CRISPR-Cas9 nucleases. Nature Reviews Genetics. 17 (5), 300-312 (2016).
- Hoban, M. D., Cost, G. J., et al. Correction of the sickle cell disease mutation in human hematopoietic stem/progenitor cells. Blood. 125 (17), 2597-2604 (2015).
- Simeonov, D. R., Gowen, B. G., et al. Discovery of stimulation-responsive immune enhancers with CRISPR activation. Nature. , (2017).
- Hultquist, J. F., Schumann, K., et al. A Cas9 ribonucleoprotein platform for functional genetic studies of HIV-host interactions in primary human T cells. Cell Reports. 17 (5), 1438-1452 (2016).
- Paix, A., Wang, Y., et al. Scalable and versatile genome editing using linear DNAs with microhomology to Cas9 sites in Caenorhabditis elegans. Genetics. 198 (4), 1347-1356 (2014).
- Lee, K., Mackley, V. A., et al. Synthetically modified guide RNA and donor DNA are a versatile platform for CRISPR-Cas9 engineering. eLife. 6, (2017).
- Minkenberg, B., Wheatley, M., Yang, Y. CRISPR/Cas9-enabled multiplex genome editing and its application. Progress in Molecular Biology and Translational Science. 149, 111-132 (2017).
- Doench, J. G., Fusi, N., et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nature Biotechnology. 34 (2), 184-191 (2016).
- Doench, J. G., Hartenian, E., et al. Rational design of highly active sgRNAs for CRISPR-Cas9-mediated gene inactivation. Nature Biotechnology. , 1-8 (2014).
- Liu, H., Wei, Z., Dominguez, A., Li, Y., Wang, X., Qi, L. S. CRISPR-ERA: a comprehensive design tool for CRISPR-mediated gene editing, repression and activation. Bioinformatics (Oxford, England). 31 (22), 3676-3678 (2015).
- Wu, X., Scott, D. A., et al. Genome-wide binding of the CRISPR endonuclease Cas9 in mammalian cells. Nature Biotechnology. 32 (7), 670-676 (2014).
- Bogenhagen, D. F., Brown, D. D. Nucleotide sequences in Xenopus 5S DNA required for transcription termination. Cell. 24 (1), 261-270 (1981).
- Cozzarelli, N. R., Gerrard, S. P., Schlissel, M., Brown, D. D., Bogenhagen, D. F. Purified RNA polymerase III accurately and efficiently terminates transcription of 5S RNA genes. Cell. 34 (3), 829-835 (1983).
- Chen, B., Gilbert, L. A., et al. Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell. 155 (7), 1479-1491 (2013).
- Gagnon, J. A., Valen, E., et al. Efficient mutagenesis by Cas9 protein-mediated oligonucleotide insertion and large-scale assessment of single-guide RNAs. PLoS ONE. 9 (5), 98186 (2014).
- Ren, X., Yang, Z., et al. Enhanced specificity and efficiency of the CRISPR/Cas9 system with optimized sgRNA parameters in Drosophila. Cell Reports. 9 (3), 1151-1162 (2014).
- Ran, F. A., Hsu, P. D., Wright, J., Agarwala, V., Scott, D. A., Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nature Protocols. 8 (11), 2281-2308 (2013).
- Serano, J. M., Martin, A., et al. Comprehensive analysis of Hox gene expression in the amphipod crustacean Parhyale hawaiensis. Developmental Biology. 409 (1), 297-309 (2016).
- Sternberg, S. H., Redding, S., Jinek, M., Greene, E. C., Doudna, J. A. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature. , 1-17 (2014).
- Lee, K., Conboy, M., et al. Nanoparticle delivery of Cas9 ribonucleoprotein and donor DNA in vivo induces homology-directed DNA repair. Nature Biomedical Engineering. 1 (11), 889-901 (2017).
