

Summary
Contamination pendant le séquençage génomique des organismes microscopiques demeure un grand problème. Ici, nous montrons une méthode pour séquencer le génome d’un tardigrade un seul spécimen avec aussi peu que 50 pg d’ADN génomique sans amplification du génome entier pour minimiser le risque de contamination.
Abstract
Tardigrades sont des animaux microscopiques qui entrent dans un état d’ametábolos appelé anhydrobiose face à la dessiccation et peuvent revenir à leur état d’origine lorsque l’eau est fournie. Le séquençage génomique des animaux microscopiques tels que la contamination bactérienne de risques tardigrades qui conduit parfois à des interprétations erronées, par exemple, au sujet de l’étendue du transfert horizontal de gènes chez ces animaux. Ici, nous fournissons une méthode d’entrée ultra basse pour séquencer le génome du tardigrade, Hypsibius dujardini, un seul spécimen. En employant l’exclusion de lavage et contaminant rigoureuse avec une extraction efficace de 50 ~ 200 pg d’ADN génomique d’un seul individu, nous avons construit une bibliothèque séquencée avec un instrument de séquençage de l’ADN. Ces bibliothèques ont été hautement reproductible et impartiale, et une analyse informatique des lectures séquencés avec autres génomes H. dujardini a montré une quantité minime de contamination. Cette méthode peut être appliquée à des tardigrades impossibles qui ne pourraient pas être séquencés à l’aide des méthodes précédentes.
Introduction
Tardigrades sont des animaux microscopiques peuvent entrer dans un état d’ametábolos appelé anhydrobiose, face à la dessiccation. Ils récupèrent de l’absorption de l’eau1,2. Dans l’état d’ametábolos, les tardigrades sont capables de tolérer des divers environnements extrêmes, incluent des températures extrêmes pressions et3 4,5, une dose élevée de lumière ultraviolette6, rayons x et rayons gamma 7 , 8et9de l’espace cosmique. Données génomiques sont un fondement indispensable pour l’étude des mécanismes moléculaires d’anhydrobiose.
Les tentatives précédentes pour séquencer le génome de tardigrades ont montré des signes de contamination bactérienne10,11,12,13,14. Séquençage génomique de ces petits organismes nécessite un grand nombre d’animaux et est sujets à une contamination bactérienne ; donc, nous avons déjà établi un protocole de séquençage en utilisant une méthode de saisie ultra basse à partir d’un seul spécimen de tardigrade, pour minimiser les risques de contaminations15. En utilisant ces données, nous avons effectué davantage une renumérotation de haute qualité et le remontage du génome de H. dujardini16,17. Ici, nous décrivons en détail cette méthode de séquençage génomique d’un seul individu tardigrade ()Figure 1). La validation de cette méthode de séquençage est au-delà de l’objectif de ce document et a déjà été abondamment discutée dans notre précédent rapport16.
Cette méthode se compose de deux parties : l’isolement d’un tardigrade unique avec le plus bas possible de la contamination et l’extraction de la qualité des niveaux du pictogramme de l’ADN. Le tardigrade est affamé et rincée avec l’eau, mais aussi des antibiotiques et observé au microscope avec un grossissement de X 500 à garantir l’élimination de toute contamination bactérienne. Les mesures et les estimations précédentes montrent qu’un seul individu de tardigrade contienne environ 50-200 pg de génomique DNA16, qui en est extraite par craquage de l’exosquelette de chitine par cycles de gel-dégel ou par homogénéisation manuelle. Cet ADN génomique est soumis à la construction de la bibliothèque et séquencé sur un instrument de séquençage de l’ADN. Une analyse informatique supplémentaire montre séquençage de haute qualité, mais aussi de faibles niveaux de contamination par rapport aux précédents projets de séquençage tardigrade.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. préparation
- Préparer un gel d’agarose de 2 % avec de l’eau distillée (DW) comme solvant dans une boîte de Petri en plastique de 90 mm et 10 mL d’un cocktail avec DW 1 % pénicilline/streptomycine. Le gel peut être conservé pendant 2 à 3 semaines dans un incubateur à 18 ° C.
NOTE : Éviter tout stockage du gel inférieures à 10 ° C, pour la basse température va diminuer le gel d’agarose, résultant dans un espace minuscule entre le gel et le mur plat de culture dans laquelle les tardigrades peuvent être pris au piège.
2. préparation des échantillons et Exclusion de Contaminant
- Recueillir un tardigrade unique, placez-le sur la gélose préparés et laver 2 x - 3 x avec DW pour éliminer les particules restantes.
- Incuber le tardigrade à 18-22 ° C pendant 24 h pour enlever tout excès de nourriture des intestins.
- Placez le tardigrade affamée en antibiotiques pénicilline/streptomycine pour 2 à 6 h enlever toute contamination bactérienne et de placer l’animal décontaminé sur un verre de lame propre à l’aide d’une pipette de P10.
- Observez le tardigrade au microscope au grossissement de X 500 et confirmer qu’il n’y a pas de bactéries restantes.
Remarque : Le fort grossissement avec un stéréomicroscope est optimal, mais un microscope optique peut être utilisé alternativement sans appliquer un lamelle couvre-objet. - Recueillir l’individu à l’aide d’une pipette de P10 avec un maximum de 5 µL de liquide, placez-le dans un tube à faible liaison polymérase chain reaction (PCR) et enlever le liquide en excès autant que possible.
Remarque : Les microtubes, tubes PCR et des pointes de pipette doivent tous être faible liaison pour minimiser la perte de l’ADN.
Remarque : Effectuer une homogénéisation dès que possible, à la dessiccation rapide ou mort de l’animal peut endommager considérablement de l’ADN génomique.
3. l’homogénéisation et l’Extraction d’ADN
- Homogénéiser l’animal pour obtenir son ADN génomique (ADNg) avec l’une des méthodes suivantes.
Remarque : Il est essentiel d’utiliser le kit spécifié montré dans la Table des matières dans les étapes d’extraction ADN suivantes, pour les autres kits (soit colonne et perle basé) ne sont pas efficaces à cette entrée extrêmement faible. Le tampon de lyse génomique a été livré avec 0,5 % bêta-mercaptoéthanol avant de l’utiliser.- Homogénéiser l’animal avec des cycles de gel-dégel,18.
- Immédiatement après étape 2.5, ajouter 100 µL de tampon de lyse dans le tube PCR contenant le tardigrade.
- Placer le tube PCR dans l’azote liquide pendant 10 min et, pendant 10 min, déplacer vers un bloc chauffant chauffé à 37 ° C. Répétez cette étape 2 x.
Remarque : Cette étape peut être répétée lorsque l’homogénéisation ne suffit pas ou peut être réalisée après le broyage manuel.
- Manuellement, écraser l’animal.
- Immédiatement après l’étape 2.5, sous un stéréomicroscope, écraser l’individu avec la pointe de P10 de la pipette en appuyant sur l’animal contre la paroi du tube PCR et immédiatement ajouter 100 µL de tampon de lyse.
Remarque : Il est essentiel de respecter cette procédure sous un stéréomicroscope, parce que le tardigrade peut facilement éclipser et inefficace de concassage se traduira par un échec pour extraire l’ADNg. Assurez-vous que la cuticule tardigrade est cassée afin que le tampon de lyse peut s’infiltrer dans l’organisme.
- Immédiatement après l’étape 2.5, sous un stéréomicroscope, écraser l’individu avec la pointe de P10 de la pipette en appuyant sur l’animal contre la paroi du tube PCR et immédiatement ajouter 100 µL de tampon de lyse.
- Homogénéiser l’animal avec des cycles de gel-dégel,18.
- Incuber le tube pendant 30 min à température ambiante pour la lyse de se produire.
Remarque : Il faut un minimum de 30 min pour une lyse efficace, mais l’incubation peut être plus longue. - Transférer le volume complet (100 µL) du mélange lyse à un microtube de liaison faible propre 1,5 mL.
- Ajouter 100 µL de tampon de lyse dans le tube PCR de liaison faible qui a été utilisé pour l’homogénéisation et est maintenant vide et après avoir effectué le mélange, transférez-le à faible liaison microtube 1,5 mL utilisé à l’étape 3.3. Répétez cette étape 2 x.
- Comme à l’étape 3.4, ajouter 300 µL de tampon de lyse dans le tube PCR de liaison faible, et, après avoir effectué, passer le mélange à basse-liaison microtube 1,5 mL. Microtube 1,5 mL faible liaison doit contenir 600 µL du mélange après cette étape.
Remarque : Ces étapes sont pour minimiser la perte de l’ADNg lié à la paroi du tube PCR, en lavant l’échantillon plusieurs fois. - Additionnez le total de 600 µL du mélange de la lyse de la colonne placée dans un tube de prélèvement et il centrifuger à 10 000 x g pendant 1 min.
- Réappliquez le débit à travers la colonne et il centrifuger à 10 000 x g pendant 1 min.
Remarque : Cette étape est essentielle pour assurer que la majeure partie de l’ADNg est lié à la colonne. - Ajouter 500 µL de tampon de lavage à la colonne et il centrifuger à 10 000 x g pendant 1 min. transfert de la colonne à un microtube 1,5 mL propre.
- Appliquer 20 µL de 10 mM Tris-HCl, pH 8,5, à la colonne, attendre 5 min à température ambiante et il centrifuger à 10 000 x g pendant 1 min.
Remarque : La mémoire tampon d’élution ne doit pas contenir de EDTA, car il interfère avec les enzymes de préparation de bibliothèque. - Ré-appliquez le flux par le biais de la colonne et après 5 min d’incubation à température ambiante, il Centrifuger pendant 1 min à 10 000 x g.
Remarque : Cette étape est essentielle pour assurer l’élution maximale de l’ADNg lié à la colonne.
4. Bibliothèque Construction séquence
-
Fragmentation de l’ADN
- Transférer 15 µL de l’éluant ADNg dans un microtube 15 µL de fragmentation de l’ADN et centrifuger le tube pendant 1 min à l’aide d’une micro-centrifugeuse sur table.
Remarque : Le microtube spécifié indiqué dans la Table des matières est optimal pour un apport réduit. La fragmentation avec faible volume sonication est essentielle, et cela ne peut pas être substitué par fragmentation enzymatique en raison de la très faible concentration d’ADN. - Fragmenter l’ADNg à 550 bp.
Remarque : Les paramètres bp 550, nous avons utilisé sont comme suit : puissance incidente de crête = 30 W, facteur d’utilisation = 20 %, cycles par burst = 50, durée du traitement = 23 s. - Après une minutieuse pipetage, transférer 10 µL du mélange ADN fragmenté dans un tube PCR de propre liaison faible.
NOTE : L’expérience peut être arrêtée ici. Préserver l’ADN à 4 ° C ou -20 ° C.
- Transférer 15 µL de l’éluant ADNg dans un microtube 15 µL de fragmentation de l’ADN et centrifuger le tube pendant 1 min à l’aide d’une micro-centrifugeuse sur table.
-
Séquence de construction de bibliothèque
Remarque : Il est absolument essentiel d’utiliser le kit spécifié dans la Table des matières dans les procédures suivantes, en raison de la faible quantité d’ADN d’entrée.- Ajouter 2 µL de tampon modèle préparation et 1 µL de l’enzyme de préparation modèle et mélanger soigneusement avec une pipette.
- Effectuer la réaction de préparation de modèle sur un thermocycleur dans les conditions suivantes : 22 ° C pendant 25 minutes, 55 ° C pendant 20 min, attente de 4 ° C et un couvercle chauffé à 101-105 ° C. Une fois que la réaction est terminée, passez à l’étape suivante.
- Ajouter 1 µL de la mémoire tampon de synthèse Bibliothèque et 1 µL de l’enzyme de synthèse de bibliothèque pour le produit de réaction de préparation modèle et incuber le mélange à 22 ° C pendant 40 min (avec une cale de 4 ° C). Une fois que la réaction est terminée, passez à l’étape suivante.
- Ajouter 30 µL de bibliothèque amplification master mix (25 µL de la mémoire tampon l’amplification dans la bibliothèque, 1 µL de l’enzyme d’amplification de bibliothèque et 4 µL d’eau exempte de nucléase) et 5 µL de réactif d’indexation.
- Mélanger le tout soigneusement avec une pipette et centrifuger le mélange brièvement avec une centrifugeuse de table.
- Effectuer un PCR avec les conditions indiquées au tableau 1.
| Température | Temps | Cycles |
| 72 ° C | 3 minutes | |
| 85 ° C | 2 minutes | |
| 98 ° C | 2 minutes | |
| 98 ° C | 20 secondes | 4 cycles |
| 67 ° C | 20 secondes | |
| 72 ° C | 40 secondes | |
| 98 ° C | 20 secondes | 16 cycles |
| 72 ° C | 50 secondes | |
| 4 ° C | Maintenez |
Tableau 1 : Conditions PCR.
-
Purification des produits PCR
Remarque : Cette étape peut être substituée avec d’autres méthodes de purification. Si une méthode alternative est utilisée, assurez-vous de vérifier la pureté de l’ADN résultant à l’aide d’un spectrophotomètre. Ratios d’absorbance de 260/280 et 260/230 doivent tous deux être au-dessus de 1,8.- Ajouter 50 µL de billes magnétiques, déposer de la solution 10 x et il a centrifuger brièvement avec une micro-centrifugeuse sur table.
- Incuber la solution pendant 2 min à température ambiante.
- Il incuber sur un support magnétique pendant 5 min ou jusqu'à ce que la solution devienne complètement clair et éliminer le surnageant.
- Ajouter 200 µL d’éthanol à 80 % fraîchement préparés dans le tube PCR de liaison faible sur un support magnétique, attendre 30 s, puis enlever le surnageant. Répétez cette étape 2 x. Ne pas déranger les perles.
- En bref, centrifuger le tube de PCR de liaison faible avec une centrifugeuse de table et enlever tout excès d’éthanol sur le support magnétique. Sécher les perles mais éviter un séchage excessif.
- Remettre en suspension les perles avec 15 µL de 10 mM Tris-HCl, pH 8,5, soigneusement déposer la solution afin que les billes magnétiques sont distribués de façon homogène, incuber le tube pendant 2 min à température ambiante et après une brève centrifugation, il incuber sur un support magnétique pour 2 min, jusqu'à ce que la solution est claire.
Remarque : La mémoire tampon d’élution ne doit pas contenir de EDTA, car il interfère avec la chimie de séquençage. - Transférer le surnageant, sans déranger le culot, dans un nouveau tube PCR de liaison faible.
NOTE : L’expérience peut être arrêtée ici. Préserver l’ADN à 4 ° C ou à-20 ° C pour le stockage de longue durée.
5. quality Check, la Quantification et la séquence de l’ADN
Remarque : La vérification de qualité n’est pas effectuée avant cette mesure en raison de la faible quantité d’ADN.
-
Validation de la distribution de taille de bibliothèque ADN
Remarque : Les autres systèmes d’électrophorèse en haute sensibilité avec un rapport numérique de la distribution peuvent être utilisés.- Retourner le réactif de tampon d’électrophorèse et gel cartouche (25-1 000 bp dans la gamme) à température ambiante.
- Ajouter 3 µL de réactif de tampon d’électrophorèse avec 1 µL de la bibliothèque de séquençage et mélangez-les soigneusement pendant 1 min avec un vortex et les centrifuger brièvement avec une centrifugeuse de table.
- Procéder à l’électrophorèse et valider la granulométrie de bibliothèque avec le logiciel associé. Le pic principal fragment doit être largement allant d’environ 300 à 1 000 bp.
-
Quantification de l’ADN
Remarque : Autres méthodes axées sur la fluorescence ou la PCR quantitative peut également être utilisé, mais spectrophotométrie devrait être évitée, car il n’est pas assez précis pour quantifier les bibliothèques de séquençage.- Ajouter 796 µL de solution tampon et 4 µL de réactif fluorescent et mélangez-les soigneusement. Diluer 190 µL de la solution de travail à deux tubes de dosage avec 197 µL à un tube d’essai.
- Ajouter 10 µL de normes avec des concentrations connues de l’ADN de chaque tube à essai contenant 190 µL de la solution de travail et 3 µL de la bibliothèque de prête dans le tube de dosage contenant 197 µL de la solution de travail.
- Vortex les tubes brièvement et centrifuger à eux sur une centrifugeuse de table. Quantifier l’ADN à l’aide d’un fluorimètre avec réglages 3 µL.
-
Séquence de bibliothèque d’ADN
Remarque : La plate-forme de séquençage doit être compatible avec le kit de construction de la bibliothèque spécifiée.- Préparer la bibliothèque de séquençage basée sur le protocole du fabricant.
- La valeur de la cellule de cassette et le débit du réactif dans l’instrument de séquençage et entrez le séquençage, exécutez informations suivant le protocole du fabricant.
- Exécutez le séquençage.
NOTE : Nous avons mené deux séries de séquençage : un échantillon/exécuter, ainsi que quatre échantillons multiplexés en un seul passage.
6. calcul analyse
- Base-appel et, le cas échéant, démultiplexer les lectures.
- Valider la qualité des données de séquence avec FastQC19.
Remarque : Pour une validation plus approfondie des données obtenues, consultez notre précédent rapport16.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Exclusion de contaminant :
Ce protocole implique un lavage complet de la Tardigrada et une stérilisation avec un traitement d’antibiotiques pour minimiser la contamination. Elle comporte également un processus de vérification visuel pour s’assurer de la conformité de ces processus. Une image microscopique faite lors de la validation (étape 2.4 du protocole) est illustrée à la Figure 2. Lorsque observé à un grossissement de X 500, les cellules bactériennes peuvent être considérées comme petites particules qui se déplacent autour le tardigrade individuel.
Validation de la qualité de bibliothèque d’ADN :
Le montant total de la bibliothèque d’ADN-Seq construite est environ 109,5 ng (7,3 ng/µL x 15 µL)16. Pour valider la distribution de la longueur de la fragmentation, un modèle d’électrophorèse doit être semblable à Figure 3. Comme nous avons mis la taille de la fragmentation à 550 PB avec un système de cisaillement de l’ADN, la bibliothèque doit être bp 550-600, y compris les adaptateurs de séquençage. On observe que la majorité de la bibliothèque de la séquence est contenue entre bp 200-1000 et est cohérente entre les répétitions (N1 - N4).
Analyse de séquence de données :
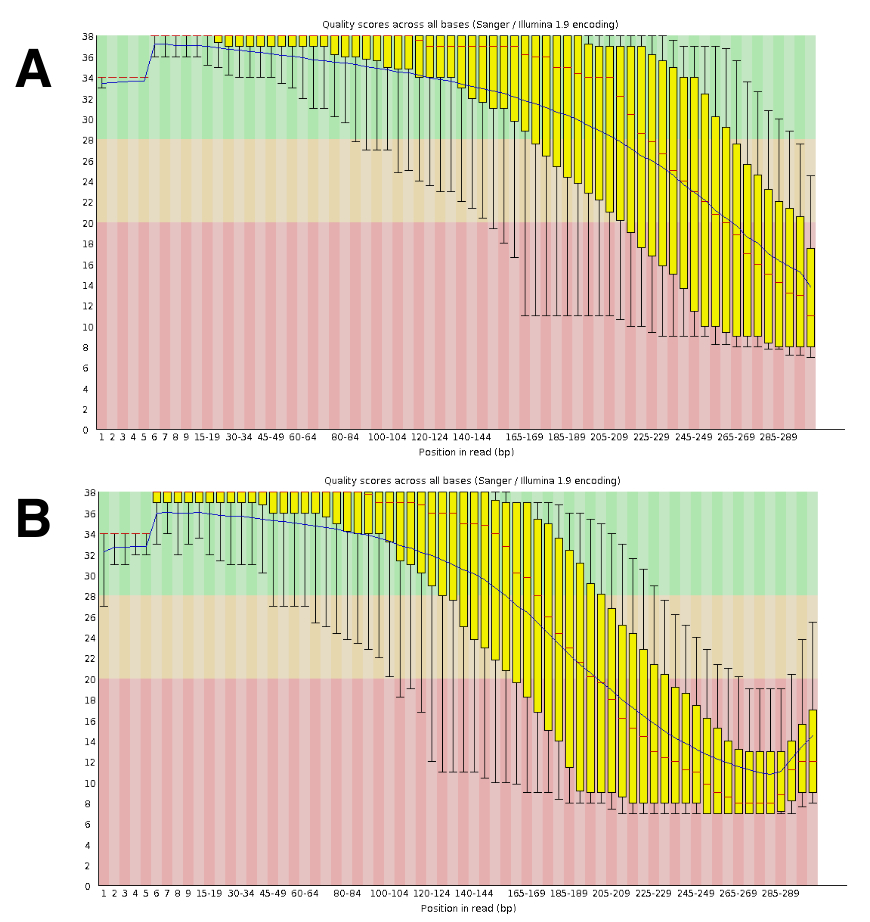
Le séquençage de l’ADN a généré environ 20 à 25 M lectures appariés par course. La validation de la qualité a été réalisée à l’aide de FastQC (Figure 4). La répartition de la qualité le long de la lecture séquencée est typique d’une série de paire de 300 PB.

Figure 1: flux de travail du présent protocole. Cette figure montre un résumé du présent protocole. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.

Figure 2 : Photo représentative d’un tardigrade sans bactéries. Cette figure montre des images d’un tardigrade (gauche) contaminé et nettoyé (à droite) tardigrade (Hypsibius dujardini), ainsi que d’autres images grossies (en bas). Cellules en forme de bâtonnet autour le tardigrade sont des contaminants et sont indiqués par une flèche. La barre d’échelle indique 100 µm. s’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.

Figure 3 : Validation de la distribution de longueur de fragment de la bibliothèque d’ADN construite. Ce panneau montre la répartition de la taille de bibliothèque de séquençage. Les lignes violettes et vertes indiquent les marqueurs supérieurs et inférieurs à 1 500 et 25 PB, respectivement. L = échelle, S = 1 échantillon/course, N1 - N4 = 4 répétitions/exécuter. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.

Figure 4 : Exemple de la validation de la qualité de l’ADN-Seq avec FastQC. ADN-Seq données ont été fournies à FastQC pour valider la performance de la séquence. Un résultat représentatif DRR055040 par la qualité de la séquence de base est affiché (lire la séquence du DDBJ Archive DRA00445516). (A), ce panneau indique le lit vers l’avant (R1). (B), ce panneau affiche les lectures inverses (R2). S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
La contamination bactérienne constitue une menace pour le séquençage génomique des organismes microscopiques. Alors que des études antérieures sur le séquençage du génome tardigrade ont filtré de contamination en utilisant une vaste informatique méthodes12,20, ils ont séquencé le génome d’un seul individu pour minimiser les risques de contaminations. Comme un tardigrade individuel contient environ 50-200 pg d’ADN génomique16 et soit revêtu d’une couche épaisse de l’exosquelette de chitine, l’exclusion des contaminants et d’extraction d’ADN de haute qualité sont les points critiques dans le présent protocole. Les cultures de tardigrades existantes ne sont pas aseptiques et ceux prélevés les sauvage portent un grand nombre de contaminants sur la surface, ainsi que les restes de nourriture dans leurs intestins. Précédents projets de séquençage du génome de tardigrades ont séquencé 10 000-100 000 individus collectivement comme un échantillon12,14, ce qui signifie que les résultats sont très susceptibles d’être influencées par les contaminants bactériens. Dans leur rapport, Boothby et al. recueillis H. dujardini particuliers à l’aide de leur comportement de phototaxie négative14, et le groupe n’a pas utilisé toutes les méthodes anti-bactérien.
Pour examiner visuellement s’il existe des contaminants, nous incubé le tardigrade en antibiotiques (pénicilline/streptomycine) et examiné l’individu sous un microscope X 500. En isolant un seul individu et inspecter soigneusement pour tous les contaminants, on réduit au minimum le risque de contamination possible. Faibles niveaux de contamination ont été confirmés d’après les données de séquençage bien16. En ce qui concerne l’extraction de l’ADN, nous avons utilisé homogénéisation manuelle, ainsi que l’homogénéisation thermique18. En soumettant l’individu tardigrade à azote liquide et à 37 ° C, les fissures ont été induites dans l’exosquelette de chitine, et le tampon de lyse a pu pénétrer dans le corps et lyser les cellules. Quand le rendement d’ADN reste moins élevé que prévu, homogénéisation thermique et manuelle peut être menée afin de maximiser le rendement.
La méthode indiquée dans cet article présente plusieurs limites. Tout d’abord, homogénéisation par cycles de gel-dégel a été appliquée d’une étude sur les nématodes ; ainsi, la méthode ne peut efficace contre ecdysozoa. Deuxièmement, en raison de l’amplification de fragments d’ADN au cours de la phase de bibliothèque de séquence de l’ADN, la possibilité d’erreurs de la PCR ne peut être ignorée. Ainsi, les données de séquence ne sont pas recommandées pour l’analyse qui nécessite des lectures de haute précision (c.-à-d., analyse SNP). En outre, comme nous l’avons dit dans le protocole, l’utilisation du kit SNA-Seq spécifié indiqué dans la Table des matières est absolument essentielle, en raison de la faible quantité d’ADN d’entrée. Ce kit de construction de bibliothèque ADN ligates séquences adaptateur Illumina avant l’amplification ; par conséquent, impossible d’appliquer cette bibliothèque pour le séquençage de long à lire en utilisant la technologie PacBio ou Nanopore. Enfin, un contrôle de la qualité de la bibliothèque d’ADN construite au cours de ce protocole se produit qu’une seule fois, après la construction de bibliothèque de séquençage. C’est en raison de l’entrée basse de l’ADN depuis plupart quantification de l’ADN et les méthodes d’électrophorèse ne peut pas détecter le 50-200 pg d’ADN. Par conséquent, nous avons effectué des contrôles de qualité, telles que l’électrophorèse (Figure 1) et basés sur la fluorescence quantitatives, qu’après l’amplification par PCR.
Un examen approfondi des analyses bioinformatiques de ces données dépasse la portée du présent article ; Cependant, nous avons brièvement déclaré plusieurs analyses que nous avons menées. Un contrôle de qualité des données de séquençage avec FastQC19 calcule les qualités par-base, duplication de séquences, des données de séquence etc. qui ont été validées peuvent être soumises à l’Assemblée de génome. Nous avons assemblé un génome 132 Mo avec MaSuRCA v3.1.321 et ont comparé les statistiques de cartographie calculées avec BWA22 et QualiMap23 de cette bibliothèque de séquençage de l’ADN avec autre H. dujardini génome assemblées16. En outre, nous aussi ont utilisé ces données de séquençage de l’ADN pour l’exclusion des contaminants dans notre étude17et ont observé que les lectures séquencées sont réparties uniformément dans tout le génome.
La plupart des projets sur les organismes non-modèle partent cultiver assez matériel de l’échantillon, comme ce fut le cas avec les tardigrades24. Les progrès techniques dans les techniques de culture ont permis à des quantités élevées de la culture tardigrade, mais les méthodes actuelles de culture ne sont pas encore aseptiques, et puisque la plupart des tardigrades sont toujours impossibles dans les laboratoires, il a été presque impossible à réaliser du génome ou séquençage du transcriptome. Cette méthode de séquençage de l’ADN d’un seul individu permet d’analyser des espèces rares tardigrade, y compris des espèces marines qui ont été étudiés en moins. En effectuant la génomique comparée à une plus large zone phylétique, mieux comprendre des mécanismes d’anhydrobiose dans tardigrades peut être réalisé.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Les auteurs n’ont rien à divulguer.
Acknowledgments
Les auteurs remercient Nozomi Abe, Yuki Takai et Nahoko Ishii pour leur soutien technique dans le séquençage génomique. Ce travail a été soutenu par la subvention pour la société japonaise pour la Promotion of Science (JSPS) Research Fellow, KAKENHI subvention des jeunes scientifiques (No.22681029) et KAKENHI subvention pour la recherche scientifique (B), n° 17 H 03620 de la JSPS, par un Subventions pour projets de recherche base scientifique de la Fondation Sumitomo (No.140340) et en partie par le Fonds de recherche du gouvernement Yamagata de la préfecture et ville de Tsuruoka, Japon. Chlorella vulgaris utilisé pour nourrir les tardigrades était une gracieuseté de Chlorella Industry Co. Ltd.
Materials
| Name | Company | Catalog Number | Comments |
| SZ61 microscope | OLYMPUS | ||
| BactoAgar | Difco Laboratories | 214010 | |
| Penicillin Streptomycin (10,000 U/mL) | Gibco by life technologies | 15140-148 | |
| VHX-5000 System | Keyence | ||
| 0.2mL Silicone coating tube | Bio Medical Science | BC-bmb20200 | |
| Quick-DNA Microprep Kit | ZYMO Research | D3021 | Use of this kit is absolutey critical; see step 3.1 |
| 1.5 mL microtube | greiner bio-one | 616-201 | See 4.1.1 |
| HIgh speed refrigerated micro centrifuge | TOMY | MX-307 | |
| Covaris M220 | Covaris Inc. | 4482277 | |
| ThruPLEX DNA-Seq kit | Rubicon Genomics | CAT. NO. R400406 | Use of this kit is absolutey critical; see step 4.2 |
| Thermal Cycler | Bioer Technology | TC-96GHbC | |
| AMPure XP reagent | BECKMAN COULTER Life Science | A63881 | |
| Ethanol | Wako | 054-027335 | |
| EB buffer | QIAGEN | 19086 | |
| 2200 TapeStation | Agilent | G2965AA | |
| D1000 Reagents | Agilent | 5067-5583 | |
| D1000 ScreenTape | Agilent | 5067-5582 | |
| Qubit dsDNA BR Buffer/Reagent | ThermoFisher Scientific | Q32850 | |
| Cubee Mini-Centrifuge | RecenttecGenereach | R5-AQBD01aqbd | |
| MiSeq 600 cycle v3 | Illumina Inc. | MS-102-3003 | |
| MiSeq Sequencer | Illumina Inc. | SY-410-1003 |
References
- Crowe, J. H., Hoekstra, F. A., Crowe, L. M. Anhydrobiosis. Annual Review of Physiology. 54 (1), 579-599 (1992).
- Mobjerg, N., et al. Survival in extreme environments - on the current knowledge of adaptations in tardigrades. Acta Physiologica. 202 (3), 409-420 (2011).
- Becquerel, P. La suspension de la vieau dessous de 1/20 K absolu par demagnetization adiabatique de L'alun de fer dans le vide les plus eléve. Comptes Rendus de l'Académie des Sciences. 231, 261-264 (1950).
- Ono, F., et al. Effect of ultra-high pressure on small animals, tardigrades and Artemia. Cogent Physics. 3 (1), 1167575 (2016).
- Horikawa, D. D., et al. Tolerance of anhydrobiotic eggs of the Tardigrade Ramazzottius varieornatus to extreme environments. Astrobiology. 12 (4), 283-289 (2012).
- Horikawa, D. D., et al. Analysis of DNA repair and protection in the Tardigrade Ramazzottius varieornatus and Hypsibius dujardini after exposure to UVC radiation. PLoS One. 8 (6), e64793 (2013).
- Horikawa, D. D., et al. Radiation tolerance in the tardigrade Milnesium tardigradum. International Journal of Radiation Biology. 82 (12), 843-848 (2006).
- May, R. M., Maria, M., Gumard, J. Action différentielle des rayons x et ultraviolets sur le tardigrade Macrobiotus areolatus, a L'état actif et desséché. Bulletin Biologique de la France et de la Belgique. 98, 349-367 (1964).
- Jonsson, K. I., Harms-Ringdahl, M., Torudd, J. Radiation tolerance in the eutardigrade Richtersius coronifer. International Journal of Radiation Biology. 81 (9), 649-656 (2005).
- Bemm, F., Weiss, C. L., Schultz, J., Forster, F. Genome of a tardigrade: Horizontal gene transfer or bacterial contamination? Proceedings of the National Academy of Sciences of the United States of America. 113 (22), E3054-E3056 (2016).
- Delmont, T. O., Eren, A. M. Identifying contamination with advanced visualization and analysis practices: metagenomic approaches for eukaryotic genome assemblies. PeerJ. 4, e1839 (2016).
- Koutsovoulos, G., et al. No evidence for extensive horizontal gene transfer in the genome of the tardigrade Hypsibius dujardini. Proceedings of the National Academy of Sciences of the United States of America. 113 (18), 5053-5058 (2016).
- Boothby, T. C., Goldstein, B., et al. Reply to Bemm et al. and Arakawa: Identifying foreign genes in independent Hypsibius dujardini genome assemblies. Proceedings of the National Academy of Sciences of the United States of America. 113 (22), E3058-E3061 (2016).
- Boothby, T. C., et al. Evidence for extensive horizontal gene transfer from the draft genome of a tardigrade. Proceedings of the National Academy of Sciences of the United States of America. 112 (52), 15976-15981 (2015).
- Arakawa, K. No evidence for extensive horizontal gene transfer from the draft genome of a tardigrade. Proceedings of the National Academy of Sciences of the United States of America. 113 (22), E3057 (2016).
- Arakawa, K., Yoshida, Y., Tomita, M. Genome sequencing of a single tardigrade Hypsibius dujardini individual. Scientific Data. 3, 160063 (2016).
- Yoshida, Y., et al. Comparative genomics of the tardigrades Hypsibius dujardini and Ramazzottius varieornatus. PLoS Biology. 15 (7), e2002266 (2017).
- He, F. Total RNA Extraction from C. elegans. Bio-protocol. Bio101, e47 (2011).
- Andrews, S. FastQC a quality-control tool for high-throughput sequence data. , http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (2015).
- Hashimoto, T., et al. Extremotolerant tardigrade genome and improved radiotolerance of human cultured cells by tardigrade-unique protein. Nature Communications. 7, 12808 (2016).
- Zimin, A. V., et al. The MaSuRCA genome assembler. Bioinformatics. 29 (21), 2669-2677 (2013).
- Li, H., Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 25 (14), 1754-1760 (2009).
- Okonechnikov, K., Conesa, A., Garcia-Alcalde, F. Qualimap 2: advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics. 32 (2), 292-294 (2016).
- Horikawa, D. D., et al. Establishment of a rearing system of the extremotolerant tardigrade Ramazzottius varieornatus: a new model animal for astrobiology. Astrobiology. 8 (3), 549-556 (2008).

{kind=link}