

Summary
This protocol provides researchers with a rapid, indirect method of measuring TLR-dependent NF-кB/AP-1 transcription factor activity in a murine macrophage cell line in response to a variety of polymeric surfaces and adsorbed protein layers that model the biomaterial implant microenvironment.
Abstract
The persistent inflammatory host response to an implanted biomaterial, known as the foreign body reaction, is a significant challenge in the development and implementation of biomedical devices and tissue engineering constructs. Macrophages, an innate immune cell, are key players in the foreign body reaction because they remain at the implant site for the lifetime of the device, and are commonly studied to gain an understanding of this detrimental host response. Many biomaterials researchers have shown that adsorbed protein layers on implanted materials influence macrophage behavior, and subsequently impact the host response. The methods in this paper describe an in vitro model using adsorbed protein layers containing cellular damage molecules on polymer biomaterial surfaces to assess macrophage responses. An NF-кB/AP-1 reporter macrophage cell line and the associated colorimetric alkaline phosphatase assay were used as a rapid method to indirectly examine NF-кB/AP-1 transcription factor activity in response to complex adsorbed protein layers containing blood proteins and damage-associated molecular patterns, as a model of the complex adsorbed protein layers formed on biomaterial surfaces in vivo.
Introduction
The foreign body reaction (FBR) is a chronic host response that can negatively impact the performance of an implanted material or device (e.g., drug delivery devices, biosensors), through the persistent release of inflammatory mediators and by impeding integration between the implanted material and the surrounding tissue1. This innate immune response is initiated by the implantation procedure and is characterized by the long-term presence of innate immune cells and fibrous capsule formation around the implant1. Within the context of material host responses, macrophage-material interactions have a significant impact on the progression of the host response and development of a FBR1. Macrophages are a diverse innate immune cell population, recruited to the implant site either from tissue-resident macrophage populations or from the blood as monocyte-derived macrophages. They begin to accumulate at the implant site shortly after implantation, and within days become the predominant cell population in the implant microenvironment. Material-adherent macrophages, along with foreign body giant cells (FBGC) formed through macrophage fusion, can persist at the material surface for the lifetime of the implant2,3. Consequently, macrophages are considered to be key players in the foreign body response due to their roles orchestrating the characteristic steps of the FBR: acute inflammatory response, tissue remodeling, and formation of fibrotic tissue1.
Toll-like receptors (TLRs) are a family of pattern recognition receptors that are expressed by many immune cells, including macrophages, and have been shown to play a significant role in inflammation and wound healing. In addition to pathogen-derived ligands, TLRs are able to bind endogenous molecules, known as damage-associated molecular patterns (DAMPs), which are released during cell necrosis and activate inflammatory signaling pathways resulting in the production of proinflammatory cytokines4. We and others have proposed that damage incurred during soft tissue biomaterial implantation procedures release DAMPs, which then adsorb to biomaterial surfaces in addition to blood proteins and modulate subsequent cell-material interactions5,6. When macrophages interact with the adsorbed protein layer on an implant, their surface TLRs may recognize adsorbed DAMPs and activate proinflammatory signaling cascades, leading to NF-κB and AP-1 transcription factor activation and production of proinflammatory cytokines. We have previously shown that murine macrophages have significantly increased NF-κB/AP-1 activity and tumor necrosis factor α (TNF-α, proinflammatory cytokine) secretion in response to DAMP-containing adsorbed protein layers on a variety of polymeric surfaces compared to surfaces with adsorbed serum or plasma only (i.e., no DAMPs present), and that this response is largely mediated by TLR2, while TLR4 plays a lesser role5.
The NF-κB/AP-1 reporter macrophage cell line (Table of Materials) used in this protocol is a convenient method to measure relative NF-κB and AP-1 activity in macrophages5,7,8. In combination with TLR pathway inhibitors, this cell line is a useful tool for investigating TLR activation and its role in inflammation in response to a variety of stimuli5,7,8. The reporter cells are a modified mouse macrophage-like cell line that can stably produce secreted embryonic alkaline phosphatase (SEAP) upon NF-κB and AP-1 transcription factor activation9. The colorimetric enzymatic alkaline phosphatase assay (Table of Materials) can then be used to quantify relative amounts of SEAP expression as an indirect measure of NF-κB/AP-1 activity. As NF-κB and AP-1 are downstream of many cell signaling pathways, neutralizing antibodies and inhibitors targeting specific TLRs (e.g., TLR2) or TLR adaptor molecules (e.g., MyD88) can be used to verify the role of a specific pathway. The methodology described in this article provides a simple and rapid approach for assessing the contribution of TLR signaling in murine macrophage responses to a variety of polymeric surfaces with adsorbed protein layers containing both blood proteins and DAMPs as an in vitro model of implanted biomaterials.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. Media and Reagent Preparation
- Prepare fibroblast media. Combine 450 mL of Dulbecco's modified Eagle medium (DMEM), 50 mL of fetal bovine serum (FBS), and 5 mL of penicillin/streptomycin. Store at 4 °C for up to 3 months.
- Prepare reporter macrophage growth media in 50 mL aliquots. Combine 45 mL of DMEM, 5 mL of FBS, 5 μg/mL mycoplasma elimination reagent (Table of Materials), and 200 μg/mL phleomycin D1 (Table of Materials). Store at 4 °C for up to 3 months.
- Prepare reporter macrophage assay media in 50 mL aliquots. Combine 45 mL of DMEM, 5 mL of heat inactivated FBS (HI-FBS), 5 μg/mL mycoplasma elimination reagent, and 200 μg/mL phleomycin D1. Store at 4 °C for up to 3 months.
2. Coating Cell Culture Surfaces with Poly(methyl methacrylate)
- Dissolve poly(methyl methacrylate) (PMMA) in chloroform at 20 mg/mL (e.g., 100 mg of PMMA in 5 mL of chloroform) in a 20 mL glass scintillation vial. Place a magnetic stir bar in the vial and allow to stir for at least 2 h, until all solids are dissolved.
CAUTION: Chloroform is harmful if inhaled. Ensure to use solvent in a fume hood while wearing PVA gloves. - Pipette 400 µL of PMMA solution onto the center of a borosilicate glass microscope slide in a spin coater, and spin at 3000 rpm for 2 min. Prepare the number of slides required for the assay, as well as 3−5 extra for water contact angle measurement. Store slides in a clean box (sprayed and wiped with 70% ethanol) for future use.
NOTE: Spin coating is often used to deposit a thin, uniform coating on a flat surface. A spin coater rotates a substrate at high speeds, using centrifugal force to spread the coating solution over the surface.- Measure water contact angle at two random positions on the surface of extra coated slides (i.e., not the slides being used for cell culture) with a goniometer to ensure glass surface was completely coated with the polymer.
NOTE: Only water of the highest purity (e.g., glass triple distilled) should be used for water contact angle measurements.
- Measure water contact angle at two random positions on the surface of extra coated slides (i.e., not the slides being used for cell culture) with a goniometer to ensure glass surface was completely coated with the polymer.
- In a biological safety cabinet (BSC) attach 8-chamber sticky wells to PMMA coated-slides using sterile forceps and following aseptic technique. Press firmly on the top of the sticky wells to make sure they are strongly attached. Incubate the slides with attached sticky-wells at 37 °C overnight to secure the seal.
- Test the seal of the sticky wells by adding 200 µL of cell culture grade (endotoxin-free) water to each well. Incubate at room temperature (RT) for 60 min and ensure no leakage before proceeding. Aspirate the water, being careful not to disturb the PMMA coating.
- Perform endotoxin-free water washes by adding 300 µL of endotoxin-free water to each well and incubating for 1 h (three times), 12 h, and 24 h prior to use to remove any remaining solvent.
- Test endotoxin concentration of the slides to be used for cell culture. Incubate 200 µL of endotoxin-free reagent water (Table of Materials) in one well of each slide for 1 h. Measure endotoxin concentration in the extract using an endpoint chromogenic endotoxin assay (Table of Materials).
NOTE: The following protocol is specific to the endotoxin assay kit listed in the Table of Materials. - Use only water and consumables (i.e., pipette tips, microcentrifuge tubes and well plates) that are certified pyrogen-free (i.e., endotoxin-free) for this work. Also, any glassware used in the preparation of the polymer-coated surfaces should be depyrogenated using dry heat sterilization (250 °C for 30 min) prior to use10. Measuring endotoxin in the extract solution, as described here, can result in an underestimation of endotoxin on the material surface11,12. Consequently, it is recommended that when developing a polymer coating protocol, perform the endotoxin assay reaction (i.e., steps 2.5.4−2.5.6 for test samples [reagent water] or spike controls) directly within wells containing the coated sample to ensure no sources of endotoxin are inadvertently introduced into the system during the coating process.
- Bring all test samples (i.e., extracts) and endotoxin assay reagents to RT. Reconstitute chromogenic reagent in assay buffer and endotoxin standard in reagent water, allow to dissolve for 5 min and gently swirl before using. Cover all bottles with paraffin film when not in use.
- Create a 5−8 point standard dilution curve of endotoxin standard ranging from the lower to the upper limit of the assay by performing a serial dilution of the endotoxin standard in reagent water.
- To control for enhancement or inhibition of the endotoxin assay in test samples, prepare a positive control (also called a spike control or spiked sample) by diluting a known amount of endotoxin in unused test sample solution.
NOTE: The concentration of the positive control should be the same concentration as a standard in the middle of the standard curve. If the recovered amount of the endotoxin spike (i.e., concentration of the positive control minus the concentration of the unspiked test sample) is within 50−200% of the nominal concentration of the endotoxin spike, the extraction solution can be considered to not significantly interfere with the assay. - Add 50 µL of standards, samples, or spike controls to each well of a 96-well plate in duplicate or triplicate. Use reagent water as a negative control.
- Add 50 µL of chromogenic reagent to every well. Add reagent quickly to all wells. Use a timer to record the amount of time it takes to add reagent to all wells. Cover the plate with an adhesive seal and incubate at 37 °C (incubation time is lot-dependent and stated on Certificate of Analysis included in the chromogenic reagent kit). Alternatively, check on the plate every 15 min during incubation until color change is observed in all standard wells.
- After incubation, add 25 µL of 50% acetic acid to each well (final concentration of 10% acetic acid per well) to stop the reaction. Add acetic acid in the same order as the chromogenic reagent was added. Read absorbance of the plate using a plate reader at 405 nm. Aspirate liquid and discard plate.
NOTE: Acetic acid addition should take the same length of time to add to each well as the chromogenic reagent took (± 30 s).
- Ultraviolet (UV) sterilize the slides for 30 min prior to cell culture experiments.
3. Coating Cell Culture Surfaces with Polydimethylsiloxane
- Mix polydimethylsiloxane (PDMS) elastomer in a 10:1 weight ratio (base:curing agent). In a biological safety cabinet, pipette approximately 10 mL of polydimethylsiloxane base into a sterile tube. Weigh the tube and slowly add curing agent until 10% has been added.
CAUTION: Use PDMS reagents in a well-ventilated area and avoid eye contact by wearing safety glasses. - Thoroughly mix the elastomer by stirring with a sterile serological pipette tip and by pipetting up and down. Add approximately 200 μL of the solution to each well of a 48-well plate. Tilt the well plate slowly to ensure complete coverage of wells with elastomer solution.
- Place the well plate with elastomer into a vacuum oven set at 50 cmHg, 40 °C. Remove the lid and cover with a single-ply wipe to prevent other debris from falling into the wells. Allow to incubate for at least 48 h.
- Confirm the wells are completely coated via visual inspection. Ensure the elastomer is fully cured by gently prodding with a sterile pipette tip before removing.
- Add 300 µL of 70% ethanol (made with absolute ethanol and endotoxin-free water) and incubate at RT for 1 h. Remove the ethanol and perform endotoxin-free water washes by adding 300 µL of endotoxin-free water to each well and incubating for 1 h (three times), 12 h, and 24 h prior to use to remove any remaining solvent.
- Incubate 200 µL of endotoxin-free water in three wells of each plate for 1 h. Measure endotoxin concentration of the water extracts using an endpoint chromogenic endotoxin assay (steps 2.5.1−2.5.6).
4. Coating Cell Culture Surfaces with Fluorinated Poly(tetrafluoroethylene)
- Make a 1 mg/mL solution of fluorinated poly(tetrafluoroethylene) (fPTFE) (e.g., add 10 mg of fPTFE to 10 mL of fluorinated solvent [Table of Materials]) in a 20 mL glass scintillation vial. Place a magnetic stir bar in the vial and allow to stir for at least 24 h, until all solids are dissolved.
- Add approximately 150 μL of the polymer solution to each well of a polystyrene 48-well plate (i.e., not tissue culture treated). Tilt the well plate slowly to ensure complete coverage of all wells with polymer solution. Replace lid.
- To ensure effective fPTFE-coating of wells, glass coverslips should be coated in fPTFE and used for water contact angle measurement (step 4.3.1). Place coverslips inside the wells of a 24-well plate. Add approximately 400 μL of the polymer solution to each well containing a coverslip. Push the coverslips down using sterile forceps, ensuring they are completely covered in polymer solution, and cover the well plate with a lid.
- Place the well plate with polymer solution and/or coverslips into a vacuum oven set at 50 cmHg, 40 °C. Remove the lid and cover with a single-ply wipe to prevent other debris from falling into the wells. Allow to incubate for at least 48 h.
- Measure water contact angle of fPTFE-coated coverslips with a goniometer to ensure effective coating.
NOTE: Only water of the highest purity (e.g., glass triple distilled) should be used for water contact angle measurements.
- Measure water contact angle of fPTFE-coated coverslips with a goniometer to ensure effective coating.
- Add 300 µL of 70% ethanol (made with absolute ethanol and endotoxin-free water) and incubate at RT for 1 h. Remove the ethanol and perform endotoxin-free water washes by adding 300 µL of endotoxin-free water to each well and incubating for 1 h (three times), 12 h, and 24 h prior to use to remove any remaining solvent.
- Incubate 200 µL of endotoxin-free water in three wells of each plate for 1 h. Measure endotoxin concentration of water extracts using an endpoint chromogenic endotoxin assay (steps 2.5.1−2.5.6).
- UV sterilize the well plates for 30 min prior to cell culture experiments.
5. Making Lysate from 3T3 Cells
- Grow 3T3 cells in multiple T150 flasks to 70% confluence. To detach cells, aspirate media, wash surface with 5 mL of PBS, and aspirate PBS. Add 5 mL of animal origin-free, recombinant cell dissociation enzyme (Table of Materials) and incubate at 37 °C for 3−5 min.
- Detach cells by gently tilting the flask back and forth. Add 5 mL of PBS to neutralize the recombinant enzyme used for cell dissociation. Transfer the detached cells from the flasks into a centrifuge tube and mix via pipetting. Perform a live cell count using a hemocytometer and cell viability dye.
NOTE: A cell dissociation enzyme that can be neutralized through dilution in PBS was selected to avoid the introduction of serum-based proteins in the lysate preparation. If trypsin is used to dissociate cells, it should be neutralized with a serum-containing solution, and an additional PBS wash should be performed to reduce the amount of serum proteins carried over into the lysate preparation. - Centrifuge the cells at 200 x g for 5 min. Aspirate the supernatant and resuspend cells in original volume (i.e., 10 mL x number of flasks) of PBS to wash off any remaining media. Repeat.
- Centrifuge the cells again at 200 x g for 5 min and aspirate the supernatant. Add the volume of PBS required to achieve a final cell concentration of 1 x 106 cells/mL. Place the cell solution into a -80 °C freezer until sample is fully frozen (at least 2 h).
- Thaw cell solution in a 37 °C water bath. Once completely thawed, place the solution back into the -80 °C freezer until totally frozen. Repeat for a total of 3 freeze-thaw cycles.
- Perform a micro bicinchoninic acid (BCA) assay on the cell lysate at a variety of dilutions (e.g., 1/100, 1/200, 1/500, 1/1000) to determine the protein concentration. Dilute the cell lysate to a protein concentration of 468.75 µg/mL, aliquot, and store at -80 °C for future use.
NOTE: Final protein concentration in a 48-well plate is 125 µg/cm2 (based on the surface area of one well, 0.75 cm2). - Perform a Western blot to assess presence of DAMPs in lysate (e.g., heat shock protein 60 [HSP60], high mobility group box 1 [HMGB1]) by loading 40−60 µg of lysate protein in loading buffer onto a 1.5 mm thick 10% polyacrylamide gel and follow standard Western blot procedures.
6. Assessing Effect of Adsorbed Protein Layers and Toll-like Receptors on NF-κB Activity of Macrophages
NOTE: For a schematic of the experimental workflow and plate layout, refer to Figure 1A and Supplemental Figure 1, respectively.
- Grow reporter macrophages in an appropriately sized flask to 70% confluence. Aspirate media, wash surface with PBS, and aspirate PBS. Add the recombinant cell dissociation enzyme and incubate at 37 °C for 8 min.
- Detach cells by firmly tapping the sides of the flask. Inactivate the recombinant cell dissociation enzyme by adding an equal volume of growth media (containing 10% FBS). Perform a live cell count using a hemocytometer and cell viability dye.
NOTE: Expected viability for the reporter macrophages following an 8 min incubation in the cell dissociation enzyme is 90%. - Centrifuge cells at 200 x g for 5 min. Aspirate supernatant and resuspend in original volume of PBS to wash cells. Centrifuge again and resuspend cells at 7.3 x 105 cells/mL in assay media (containing heat inactivated FBS).
- Separate cell suspension into 3 different tubes: TLR4 inhibitor, anti-TLR2, and untreated. Incubate cells with 1 µg/mL TLR4 inhibitor for 60 min at RT or with 50 µg/mL anti-TLR2 for 30 min at RT.
- Add 200 µL of lysate, 10% FBS, 10% commercial mouse plasma (Table of Materials), or a mixture of the protein solutions to a 48-well plate (or equivalent) and allow protein to adsorb at 37 °C for the desired amount of time (i.e., 30 min, 60 min, or 24 h). Aspirate protein solutions from wells, using a fresh Pasteur pipette for each protein solution, and wash surfaces with 250 µL of PBS for 5 min. Aspirate PBS. Repeat for a total of 3 washes.
NOTE: This step may need to be started earlier in the protocol depending on desired adsorption time. Adjust protocol accordingly. - After incubation period with the TLR4 inhibitor or anti-TLR2, pipette cells to resuspend. Add 200 µL of cell solution to each well.
- For TLR2 positive control condition, add Pam3CSK4 to a final concentration of 150 ng/mL. For TLR4 positive control condition, add lipopolysaccharide (LPS) to a final concentration of 1.5 µg/mL. Incubate cells at 37 °C for 20 h.
- Sample 20 µL of supernatant from each well and plate in duplicate into a 96-well plate. Include three wells of 20 µL assay media as a background control. Add 200 µL of SEAP reporter assay reagent to each well. Cover the plate with an adhesive seal and incubate for 2.5 h at 37 °C.
NOTE: The incubation time may vary depending on experimental conditions, and should be optimized for a strong difference in absorbance between positive and negative control wells.- Transfer the remainder of the supernatant to a 1.5 mL tube (per well). Centrifuge at 1,000 x g for 10 min to pellet any debris. Transfer supernatant to a new 1.5 mL tube and store at -80 °C. Analyze supernatant for the presence of proinflammatory cytokines (e.g., TNF-α, interleukin 6) via enzyme-linked immunosorbent assay (ELISA).
- Remove adhesive plate seal. Read absorbance of the plate using a plate reader at 635 nm. Aspirate liquid and discard plate.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Cleaning methods for the polymer-coated surfaces were tested to ensure there was no disruption of the coating, which would be seen as a change in the water contact angle to an uncoated glass coverslip (Figure 2). Soaking PMMA-coated microscope slides in 70% ethanol for 1 h was found to remove the PMMA coating (Figure 2, left panel), likely due to the solubility of PMMA in 80 wt% ethanol13, therefore PMMA-coated surfaces were cleaned using 30 min of UV sterilization alone. The concentration of PMMA for coating was optimized previously5. A 1 h 70% ethanol soak was used to clean PDMS, and UV sterilization was neglected since UV light can cause chain scission and influence the surface wetting properties of PDMS14. Both 70% ethanol soak and UV sterilization did not influence the water contact angle of fPTFE-coated coverslips (Figure 2, right panel), therefore the two methods, in succession, were used to clean fPTFE coatings. The method of fPTFE coating was previously described by the Grainger group15.
A Western blot was performed on the 3T3 lysate to ensure that DAMP species were present in the complex molecular mixture. The results showed that both HMGB1 and HSP60, two well-documented DAMPs16,17, were present in the lysate (Figure 1B). The adsorption of TLR ligands from the lysate onto the polymer surfaces was confirmed by culturing reporter macrophages (untreated, TLR2 neutralized, or TLR4 inhibited) for 20 h on protein-adsorbed polymer surfaces (i.e., tissue culture-treated polystyrene [TCPS], PMMA, PDMS, fPTFE), and then indirectly assessing NF-кB/AP-1 activity based on SEAP production using an enzymatic assay (Figure 1C and Figure 3). Furthermore, the reporter macrophages had significantly increased NF-кB/AP-1 activity on adsorbed lysate compared to adsorbed FBS or plasma and no pre-adsorbed protein (media) (Figure 4). TLR ligands synthetic triacylated lipopeptide (Pam3CSK4, TLR2 ligand) and lipopolysaccharide (LPS, TLR4 ligand) were included as positive controls to confirm the antibody or inhibitor and the assay were working properly. TLR2 neutralization had a noticeably stronger reduction in NF-кB/AP-1 response of reporter macrophages to adsorbed lysate compared to TLR4 inhibition. As well, small amounts of lysate diluted in serum (based on total protein) induced significantly increased NF-кB/AP-1 response compared to serum alone, with the lowest effective dilution dependent on the polymer surface (Figure 5). These results demonstrate the potency of the adsorbed lysate-derived molecules on inducing TLR-dependent NF-кB/AP-1 activity in reporter macrophages on a variety of polymeric surfaces.

Figure 1: Methods and results for the alkaline phosphatase assay of NF-кB/AP-1 reporter macrophages on TCPS, PMMA, PDMS, and fPTFE. (A) Diagram of the workflow for the reporter macrophage alkaline phosphatase assay. (B) Western blot of lysate confirming the presence of DAMP species HMGB1 and HSP60, with β-actin as the loading control. (C) NF-кB/AP-1 activity (represented by absorbance) of reporter macrophages cultured on media (negative control), 10% FBS, lysate, and Pam3CSK4 (TLR2 ligand, positive control) for 20 h. Data shows the results of one experiment and is representative of results from at least 2 separate experiments, shown as mean ± standard deviation (SD). Each experiment used n = 3 separate wells per condition, and each well was plated in duplicate for the enzymatic assay. Analyzed using one-way ANOVA and Tukey post-hoc test. *** p < 0.001. This figure has been adapted with permission from McKiel and Fitzpatrick5. Copyright 2018 American Chemical Society. Please click here to view a larger version of this figure.

Figure 2: Optimization of cleaning methods for PMMA- and fPTFE-coated surfaces, assessed using water contact angle (WCA). Measurements were taken on 2 separate spots of at least 3 coverslips. Data is shown as mean ± SD. Analyzed using one-way ANOVA and Tukey post-hoc test. * p < 0.05. This figure has been adapted with permission from McKiel and Fitzpatrick5. Copyright 2018 American Chemical Society. Please click here to view a larger version of this figure.

Figure 3: TLR-mediated NF-кB/AP-1 activity (represented by absorbance) of reporter macrophages cultured on 10% FBS (control), lysate, and positive control for 20 h. (A) Influence of TLR2 neutralization on reporter macrophage responses to adsorbed lysate. Positive control is Pam (Pam3CSK4, TLR2 ligand). (B) Influence of TLR4 inhibition on reporter macrophage response to adsorbed lysate. Positive control is LPS (TLR4 ligand). Data shows the results of one experiment and is representative of results from at least 2 separate experiments, shown as mean ± SD. Each experiment used n = 3 separate wells per condition, and each well was plated in duplicate for the enzymatic assay. Analyzed using one-way ANOVA and Tukey post-hoc test. ** p < 0.01, *** p < 0.001. This figure has been adapted with permission from McKiel and Fitzpatrick5. Copyright 2018 American Chemical Society. Please click here to view a larger version of this figure.

Figure 4: NF-кB/AP-1 activity (represented by absorbance) of reporter macrophages cultured on media (negative control), 30 min and 24 h adsorbed protein layers, and Pam3CSK4 (positive control) on TCPS for 20 h. Data is combined from 3 separate experiments and shown as mean ± SD. Each experiment used n = 3 separate wells per condition, and each well was plated in duplicate for the enzymatic assay (i.e., n = 9 non-independent cell culture wells and n = 18 non-independent enzymatic assay wells). Analyzed using one-way ANOVA and Tukey post-hoc test. *** p < 0.001. Please click here to view a larger version of this figure.
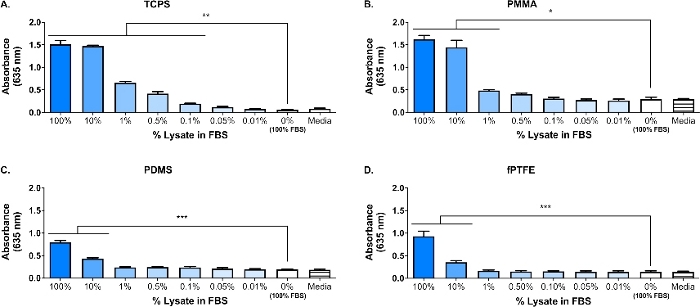
Figure 5: Reporter macrophage NF-кB/AP-1 activity (represented by absorbance) after 20 h in response to dilutions of lysate in FBS (total protein = 280 µg/well) adsorbed to polymer surfaces for 30 min. (A) TCPS. (B) PMMA. (C) PDMS. (D) fPTFE. Data shows the results of one experiment and is representative of results from at least 2 separate experiments, shown as mean ± SD. Each experiment used n = 3 separate wells per condition, and each well was plated in duplicate for the enzymatic assay. Analyzed using one-way ANOVA and Tukey post-hoc test. * p < 0.05, ** p < 0.01, *** p < 0.001. This figure has been adapted with permission from McKiel and Fitzpatrick5. Copyright 2018 American Chemical Society. Please click here to view a larger version of this figure.

Supplemental Figure 1: Example layouts used for NF-кB/AP-1 reporter macrophage cell culture assay in 8-chamber and 48-well plate formats. Please click here to view a larger version of this figure.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
A primary focus of our lab is the host response to solid biomaterial soft tissue implants, and in particular how the cellular damage incurred during the implantation procedure impacts the host response. The work presented here describes preliminary experiments using a reporter macrophage cell line and in vitro-generated DAMP-containing cellular lysate, to investigate the influence of molecules released during cellular damage (i.e., from the implant surgery) on macrophage responses to biomaterials. Fibroblast cell lysate was used to model the cellular damage and release of DAMPs due to biomaterial placement. Fibroblasts were chosen to create the lysate because of the prevalence of fibroblasts in soft tissue, as well as their ability to secrete a variety of extracellular matrix (ECM) proteins, including fibronectin18. Freeze-thaw cycling was chosen as the method of lysis to produce both intracellular and ECM-derived DAMPs, similar to what would be present in the implant environment. Protease inhibitors were not used to make this lysate. While uncontrolled cell lysis like freeze-thaw cycling can result in the release of proteases that may degrade DAMPs, these enzymes would also likely be present in the biomaterial implant environment when cells are damaged during the implantation procedure. The presence of DAMPs in the complex molecular mixture of the lysate was confirmed by Western blot (Figure 1B; HMGB1 and HSP60) and SEAP reporter assay (Figure 1C; NF-кB/AP-1 activity in response to adsorbed lysate). We have also performed assays where lysate was diluted in FBS based on total protein concentration and adsorbed onto cell culture surfaces (Figure 5) to better reflect the complexity of the implant environment, since it will contain an abundance of blood proteins as well as DAMPs6. Reporter macrophage NF-кB/AP-1 activity remained significantly increased on adsorbed layers from lysate diluted in FBS, and the lowest dilution to achieve significant activation was surface dependent, ranging from 0.1% (TCPS) to 10% (PDMS and fPTFE).
The polymers PMMA, PDMS, and PTFE were chosen for this work because they are nondegradable and have been used extensively in the literature to assess protein adsorption and macrophage response to biomaterials19,20,21,22,23,24,25. TCPS was also used for comparison since it is a common substrate used for in vitro macrophage and TLR signaling work21,26,27,28. The materials used in our work are representative examples of non-degradable, solid biomaterials. However, many other materials could be used with this model, provided the material can be coated onto cell culture plates or microscope slides and properly decontaminated. The NF-κB/AP-1 reporter macrophage cell line was selected for this in vitro model because it enables rapid, indirect measurement of NF-κB/AP-1 activity through the NF-κB/AP-1 inducible expression of SEAP. NF-κB/AP-1 reporter macrophages require the use of phleomycin D1 in the culture media as a selective antibiotic to ensure that only cells with the NF-кB/AP-1 inducible SEAP gene are present29. For the alkaline phosphatase assay, it is critical to use HI-FBS in the cell culture media to avoid potential false positive results generated by alkaline phosphatases present in serum. Our research to date suggests that FBS-adsorbed surfaces do not generate a detectable false positive result, likely because the serum molecules are strongly adsorbed to the culture surface and are not released into the supernatant. The culture time point for the reporter macrophages (20 h), assay incubation timepoint (2.5 h), and absorbance reading wavelength (635 nm) for the alkaline phosphatase assay were optimized with this system to ensure robust and reproducible measurements for all conditions.
An initial protein adsorption timepoint of 30 min was chosen for this work due to its common use in protein adsorption literature (Figure 1C)30,31,32,33,34. However, we have also explored longer adsorption times (i.e., 60 min and 24 h, Figure 4) to better represent the adsorbed protein layer that macrophages would interact with in vivo, which is likely to occur 4−24 h following implantation1. It has been postulated that the majority of protein adsorption and exchange occurs in the first 60 min of exposure to a surface26,35,36, therefore a 60 min adsorption time may be a more relevant timepoint. We have also moved from using FBS as a negative control for the presence of DAMPs in the adsorbed protein layer to commercial mouse plasma. The rationale for using plasma instead of serum is that plasma proteins are known to play significant roles in protein adsorption and macrophage response1, and that plasma provides a better representation of the proteins in the wound environment. Plasma used in protein adsorption experiments is commonly prepared as a 1−10% dilution26,36,37, which motivated our use of 10% plasma. Human plasma is commonly used26,36, as it is easier to obtain in large quantities and more clinically relevant, compared to mouse plasma. However, we chose to use commercial mouse plasma for in this model to keep the species of the protein solutions consistent with that of the reporter cells.
The use of the reporter macrophage cell line introduced some limitations within the study. First, using a murine leukemic macrophage cell line has inherent limitations, as the phenotype and behavior may vary from primary macrophage cultures. While this limitation will be addressed in future work using primary macrophages, the parental macrophage cell line was shown to closely mimic mouse bone marrow-derived macrophages in terms of their cell surface receptors and response to microbial ligands for TLRs 2, 3 and 438. Furthermore, the NF-κB/AP-1 reporter macrophages yielded similar results in response to HMGB1 and LPS stimulation when compared to peritoneal primary murine macrophages39. It should be noted that the NF-κB/AP-1 reporter macrophages, and their parental strain, do not express TLR540. Researchers have demonstrated that HMGB1 was able to activate NF-κB transcription factors via the TLR5 signaling pathways in HEK-293 cells stably transfected with human TLR541. Therefore, the contribution of HMGB1-TLR5 signaling to overall NF-κB activity on lysate-coated surfaces was neglected in this model. Additionally, the reporter macrophages and their parental strain do not express the ASC adaptor protein, and consequently do not form most types of inflammasomes and cannot process inactive IL-1β or inactive IL-18 to their mature forms42. Therefore, the model we have used does not account for the contribution of ASC-dependent inflammasome activity and subsequent autocrine IL-1β and IL-18 signaling in macrophage responses to lysate-adsorbed surfaces. Consequently, this assay is intended as a preliminary examination of TLR-dependent NF-κB activation, and subsequent research using primary macrophages is recommended to provide a more complete and representative understanding of macrophage activation and phenotype on material surfaces of interest.
The alkaline phosphatase assay indirectly measures the NF-кB/AP-1 activity of the reporter macrophages. However, there are many signaling pathways other than TLRs that involve NF-кB/AP-1 (e.g., interleukin-1 receptor [IL-1R]43 and tumor necrosis factor receptor [TNFR]44). Therefore, it was necessary to assess the contribution of TLR2 and TLR4 signaling in the increased NF-кB/AP-1 response to lysate-adsorbed surfaces using inhibition assays (Figure 3). The rationale for selecting these two surface TLRs was that at least 23 DAMPs that have been shown to signal through TLR2 and TLR445, including the well-characterized HMGB1, and both receptors are expressed on the cell surface and can interact directly with the biomaterial surface6. The TLR2 and TLR4 inhibition assays demonstrated that when TLR2 or TLR4 signaling were blocked, the NF-кB/AP-1 response of the reporter macrophages to adsorbed lysate was reduced, indicating that both pathways are involved. However, there was a noticeably larger reduction in NF-кB/AP-1 activity when TLR2 signaling was neutralized, suggesting that TLR2 may play a primary role in the response of reporter macrophages to adsorbed lysate. We recognize that there may be some off-target inhibition with the TLR signaling pathway neutralizing antibodies and inhibitors. A neutralizing antibody was used to inhibit the TLR2 pathway since there were not commercially available TLR2 inhibitor molecules at the time of this work.
The methods presented here use lysate, as a complex source of DAMPs, and NF-κB/AP-1 reporter macrophages as an in vitro model for macrophage responses to DAMPs and other proteins adsorbed to polymeric biomaterials (Figure 1). We anticipate our protocol can be used to quickly analyze NF-κB/AP-1 responses and upstream TLR signaling of reporter macrophages to a variety of materials (including degradable materials, porous scaffolds or hydrogels) and adsorbed protein layers (Figure 3). However, the use of porous materials and hydrogels will introduce complexity within the system, as it may be challenging to distinguish between adsorbed molecules and entrained molecules. We also anticipate that this protocol can be easily adapted to investigate the contribution of other signaling pathway upstream of NF-кB/AP-1 (e.g., C-type lectin receptors46 and nucleotide-binding oligomerization domain (NOD)-like receptors47) with the appropriate inhibitors. Furthermore, the NF-κB/AP-1 response of reporter macrophages could be compared between different materials, provided responses are normalized to baseline cell activity (i.e., cells in media on each surface with no pre-adsorbed protein) and all materials have undetectable endotoxin levels.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
The authors have nothing to disclose.
Acknowledgments
The authors gratefully acknowledge operational funding from Canadian Institutes of Health Research Project (PTJ 162251), Queen's University Senate Advisory Research Committee and infrastructure support from the Canadian Foundation for Innovation John Evan's Leadership Fund (Project 34137) and the Ministry of Research and Innovation Ontario Research Fund (Project 34137). L.A.M. was supported by a Queen's University R. Samuel McLaughlin Fellowship, a Natural Sciences and Engineering Research Council of Canada Canadian Graduate Scholarship Master's Award and an Ontario Graduate Scholarship. The authors would like to thank Dr. Myron Szewczuk for his generous gift of the NF-κB/AP-1 reporter macrophage cell line and Drs. Michael Blennerhassett and Sandra Lourenssen for the use of their gel imaging system and plate reader.
Materials
| Name | Company | Catalog Number | Comments |
| Cell culture reagents | |||
| anti-mouse/human CD282 (TLR2) | Biolegend | 121802 | |
| CLI-095 (TLR4 inhibitor) | Invivogen | TLRL-CLI95 | |
| C57 complement plasma K2 EDTA 10ml, innovative grade US origin | InnovativeResearch | IGMSC57-K2 EDTA-Compl-10ml | Mouse plasma |
| Dulbecco's modified eagle medium (DMEM) | Sigma Aldrich | D6429-500ML | |
| Dulbecco's phosphate buffered saline (DPBS) | Fisher Scientific | 14190250 | No calcium, no magnesium |
| Fetal bovine serum (FBS), research grade | Wisent | 98150 | |
| LPS-EK | Invivogen | TLRL-EKLPS | Lipopolysaccharide from Escherichia coli K12 |
| NIH/3T3 fibroblasts | ATCC | CRL-1658 | |
| Pam3CSK4 | Invivogen | tlrl-pms | Synthetic triacylated lipopeptide - TLR1/2 ligand |
| Penicillin/streptomycin | Sigma Aldrich | P4333-100ML | |
| Plasmocin | Invivogen | ANT-MPP | Mycoplasma elimination reagent |
| RAW-Blue cells | Invivogen | raw-sp | NF-κB/AP-1 reporter macrophage cell line |
| Trypan blue solution, 0.4% | Fisher Scientific | 15250061 | |
| TrypLE express enzyme (1X) | Fisher Scientific | 12604021 | animal origin-free recombinant cell dissociation enzyme |
| Zeocin | Invivogen | ANT-ZN-1 | |
| Kits and assays | |||
| ELISA precoated plates, mouse IL-6 | Biolegend | B213022 | |
| ELISA precoated plates, mouse TNF-α | Biolegend | B220233 | |
| Endotoxin (Escherichia coli) - Control standard endotoxin (CSE) | Associates of Cape Cope Inc. | E0005-5 | Endotoxin for standard curve in chromogenic endotoxin assay |
| LAL water, 100 mL | Associates of Cape Cope Inc. | WP1001 | Used with chromogenic endotoxin assay |
| Micro BCA protein assay | Fisher Scientific | PI23235 | |
| Limulus amebocyte lysate (LAL) Pyrochrome endotoxin test kit | Associates of Cape Cope Inc. | C1500-5 | Chromogenic endotoxin assay reagent |
| QUANTI-Blue alkaline phosphatase detection medium | Invivogen | rep-qb2 | Alkaline phosphatase assay to indirectly measure NF-κB/AP-1 activity |
| Polymeric coating reagents | |||
| Chloroform, anhydrous | Sigma Aldrich | 288306-1L | |
| Ethyl alcohol anhydrous | Commercial Alcohols | P006EAAN | Sigma: Reagent alcohol, anhydrous, 676829-1L |
| Straight tapered fine tip forceps | Fisher Scientific | 16-100-113 | |
| Fluorinert FC-40 solvent | Sigma Aldrich | F9755-100ML | Fluorinated solvent for fPTFE |
| Cell culture grade water (endotoxin-free) | Fisher Scientific | SH30529LS | |
| Poly(methyl methacrylate) (PMMA) | Sigma Aldrich | 182230-25G | |
| Sylgard 184 elastomer kit | Fisher Scientific | 50822180 | |
| Teflon-AF (fPTFE) | Sigma Aldrich | 469610-1G | Poly[4,5-difluoro-2,2-bis(trifluoromethyl)-1,3-dioxole-co-tetrafluoroethylene] |
| Consumables | |||
| Adhesive plate seals | Fisher Scientific | AB-0580 | |
| Axygen microtubes, 1.5 mL | Fisher Scientific | 14-222-155 | |
| Borosilicate glass scintillation vials, with white polypropylene caps | Fisher Scientific | 03-337-4 | |
| Clear PS 48-well plate | Fisher Scientific | 08-772-52 | |
| Clear TCPS 96-well plate | Fisher Scientific | 08-772-2C | |
| Clear TCPS 48-well plate | Fisher Scientific | 08-772-1C | |
| Cover glasses, circles | Fisher Scientific | 12-545-81 | |
| Falcon tissue culture treated flasks, T25 | Fisher Scientific | 10-126-10 | |
| sticky-Slide 8 Well | Ibidi | 80828 | |
| Superfrost microscope slides | Fisher Scientific | 12-550-15 | |
| Tissue culture treated flasks, T150 | Fisher Scientific | 08-772-48 |
References
- Anderson, J. M., Rodriguez, A., Chang, D. T. Foreign body reaction to biomaterials. Seminars in Immunology. 20 (2), 86-100 (2008).
- Anderson, J. M., Miller, K. M. Biomaterial biocompatibility and the macrophage. Biomaterials. 5 (1), 5-10 (1984).
- Collier, T. O., Anderson, J. M. Protein and surface effects on monocyte and macrophage adhesion, maturation, and survival. Journal of Biomedical Materials Research. 60 (3), 487-496 (2002).
- Bianchi, M. E. DAMPs, PAMPs and alarmins: all we need to know about danger. Journal of Leukocyte Biology. 81 (1), 1-5 (2007).
- McKiel, L. A., Fitzpatrick, L. E. Toll-like Receptor 2-Dependent NF-κB/AP-1 Activation by Damage-Associated Molecular Patterns Adsorbed on Polymeric Surfaces. ACS Biomaterials Science & Engineering. 4 (11), 3792-3801 (2018).
- Babensee, J. E. Interaction of dendritic cells with biomaterials. Seminars in Immunology. 20 (2), 101-108 (2008).
- Sintes, J., Romero, X., de Salort, J., Terhorst, C., Engel, P. Mouse CD84 is a pan-leukocyte cell-surface molecule that modulates LPS-induced cytokine secretion by macrophages. Journal of Leukocyte Biology. 88 (4), 687-697 (2010).
- Tom, J. K., Mancini, R. J., Esser-Kahn, A. P. Covalent modification of cell surfaces with TLR agonists improves and directs immune stimulation. Chemical Communications. 49 (83), 9618-9620 (2013).
- Abdulkhalek, S., et al. Neu1 sialidase and matrix metalloproteinase-9 cross-talk is essential for toll-like receptor activation and cellular signaling. Journal of Biological Chemistry. 286 (42), 36532-36549 (2011).
- Gorbet, M. B., Sefton, M. V. Endotoxin: The uninvited guest. Biomaterials. 26 (34), 6811-6817 (2005).
- Xing, Z., Pabst, M. J., Hasty, K. A., Smith, R. A. Accumulation of LPS by polyethylene particles decreases bone attachment to implants. Journal of Orthopaedic Research. 24 (5), 959-966 (2006).
- Ding, H., et al. Comparison of the cytotoxic and inflammatory responses of titanium particles with different methods for endotoxin removal in RAW264.7 macrophages. Journal of Materials Science: Materials in Medicine. 23 (4), 1055-1062 (2012).
- Hoogenboom, R., Becer, C. R., Guerrero-Sanchez, C., Hoeppener, S., Schubert, U. S. Solubility and thermoresponsiveness of PMMA in alcohol-water solvent mixtures. Australian Journal of Chemistry. 63 (8), 1173-1178 (2010).
- Efimenko, K., Wallace, W. E., Genzer, J. Surface modification of Sylgard-184 poly(dimethyl siloxane) networks by ultraviolet and ultraviolet/ozone treatment. Journal of Colloid and Interface Science. 254 (2), 306-315 (2002).
- Godek, M. L., Sampson, J. A., Duchsherer, N. L., McElwee, Q., Grainger, D. W. Rho GTPase protein expression and activation in murine monocytes/macrophages is not modulated by model biomaterial surfaces in serum-containing in vitro cultures. Journal of Biomaterials Science. Polymer Edition. 17 (10), 1141-1158 (2006).
- Park, J. S., et al. Involvement of Toll-like Receptors 2 and 4 in Cellular Activation by High Mobility Group Box 1 Protein. Journal of Biological Chemistry. 279 (9), 7370-7377 (2004).
- Ohashi, K., Burkart, V., Flohé, S., Kolb, H. Cutting Edge: Heat Shock Protein 60 Is a Putative Endogenous Ligand of the Toll-Like Receptor-4 Complex. The Journal of Immunology. 164 (2), 558-561 (2000).
- Wong, T., McGrath, J. A., Navsaria, H. The role of fibroblasts in tissue engineering and regeneration. British Journal of Dermatology. 156 (6), 1149-1155 (2007).
- van Wachem, P. B., et al. The influence of protein adsorption on interactions of cultured human endothelial cells with polymers. Journal of Biomedical Materials Research. 21 (6), 701-718 (1987).
- Miller, K. M., Anderson, J. M. Human monocyte/macrophage activation and interleukin 1 generation by biomedical polymers. Journal of Biomedical Materials Research. 22 (8), 713-731 (1988).
- Bonfield, T. L., Colton, E., Anderson, J. M. Plasma protein adsorbed biomedical polymers: Activation of human monocytes and induction of interleukin 1. Journal of Biomedical Materials Research. 23 (6), 535-548 (1989).
- González, O., Smith, R. L., Goodman, S. B. Effect of size, concentration, surface area, and volume of polymethylmethacrylate particles on human macrophages in vitro. Journal of Biomedical Materials Research. 30 (4), 463-473 (1996).
- Anderson, J. M., et al. Protein adsorption and macrophage activation on polydimethylsiloxane and silicone rubber. Journal of Biomaterials Science. Polymer Edition. 7 (2), 159-169 (1995).
- Lord, M. S., Foss, M., Besenbacher, F. Influence of nanoscale surface topography on protein adsorption and cellular response. Nano Today. 5 (1), 66-78 (2010).
- Chen, S., et al. Characterization of topographical effects on macrophage behavior in a foreign body response model. Biomaterials. 31 (13), 3479-3491 (2010).
- Shen, M., Horbett, T. A. The effects of surface chemistry and adsorbed proteins on monocyte/macrophage adhesion to chemically modified polystyrene surfaces. Journal of Biomedical Materials Research. 57 (3), 336-345 (2001).
- Love, R. J., Jones, K. S. The recognition of biomaterials: Pattern recognition of medical polymers and their adsorbed biomolecules. Journal of Biomedical Materials Research Part A. 101 (9), 2740-2752 (2013).
- McNally, A. K., Anderson, J. M. Phenotypic expression in human monocyte-derived interleukin-4-induced foreign body giant cells and macrophages in vitro: Dependence on material surface properties. Journal of Biomedical Materials Research Part A. 103 (4), 1380-1390 (2015).
- Gambhir, V., et al. The TLR2 agonists lipoteichoic acid and Pam3CSK4 induce greater pro-inflammatory responses than inactivated Mycobacterium butyricum. Cellular Immunology. 280 (1), 101-107 (2012).
- Suzuki, O., Yagishita, H., Yamazaki, M., Aoba, T. Adsorption of Bovine Serum Albumin onto Octacalcium Phosphate and its Hydrolyzates. Cells and Materials. 5 (1), 45-54 (1995).
- Johnston, R. L., Spalton, D. J., Hussain, A., Marshall, J. In vitro protein adsorption to 2 intraocular lens materials. Journal of Cataract and Refractive Surgery. 25 (8), 1109-1115 (1999).
- Jin, J., Jiang, W., Yin, J., Ji, X., Stagnaro, P. Plasma proteins adsorption mechanism on polyethylene-grafted poly(ethylene glycol) surface by quartz crystal microbalance with dissipation. Langmuir. 29 (22), 6624-6633 (2013).
- Swartzlander, M. D., et al. Linking the foreign body response and protein adsorption to PEG-based hydrogels using proteomics. Biomaterials. 41, 26-36 (2015).
- Chamberlain, M. D., et al. Unbiased phosphoproteomic method identifies the initial effects of a methacrylic acid copolymer on macrophages. Proceedings of the National Academy of Sciences. 112 (34), 10673-10678 (2015).
- Dillman, W. J., Miller, I. F. On the adsorption of serum proteins on polymer membrane surfaces. Journal of Colloid And Interface Science. 44 (2), 221-241 (1973).
- Ishihara, K., Ziats, N. P., Tierney, B. P., Nakabayashi, N., Anderson, J. M. Protein adsorption from human plasma is reduced on phospholipid polymers. Journal of Biomedical Materials Research. 25 (11), 1397-1407 (1991).
- Warkentin, P., Wälivaara, B., Lundström, I., Tengvall, P. Differential surface binding of albumin, immunoglobulin G and fibrinogen. Biomaterials. 15 (10), 786-795 (1994).
- Berghaus, L. J., et al. Innate immune responses of primary murine macrophage-lineage cells and RAW 264.7 cells to ligands of Toll-like receptors 2, 3, and 4. Comparative Immunology, Microbiology and Infectious Diseases. 33 (5), 443-454 (2010).
- Zhang, Y., Karki, R., Igwe, O. J. Toll-like receptor 4 signaling: A common pathway for interactions between prooxidants and extracellular disulfide high mobility group box 1 (HMGB1) protein-coupled activation. Biochemical Pharmacology. 98 (1), 132-143 (2015).
- Mizel, S. B., Honko, A. N., Moors, M. A., Smith, P. S., West, A. P. Induction of macrophage nitric oxide production by Gram-negative flagellin involves signaling via heteromeric Toll-like receptor 5/Toll-like receptor 4 complexes. Journal of Immunology. 170 (12), 6217-6223 (2003).
- Das, N., et al. HMGB1 Activates Proinflammatory Signaling via TLR5 Leading to Allodynia. Cell Reports. 17 (4), 1128-1140 (2016).
- Pelegrin, P., Barroso-Gutierrez, C., Surprenant, A. P2X7 Receptor Differentially Couples to Distinct Release Pathways for IL-1β in Mouse Macrophage. The Journal of Immunology. 180 (11), 7147-7157 (2008).
- Tak, P. P., Firestein, G. S. NF-κB: A key role in inflammatory diseases. Journal of Clinical Investigation. 107 (1), 7-11 (2001).
- Ashkenazi, A., Dixit, V. M. Death receptors: signaling and modulation. Science. 281 (5381), 1305-1308 (1998).
- Erridge, C. Endogenous ligands of TLR2 and TLR4: agonists or assistants. Journal of Leukocyte Biology. 87 (6), 989-999 (2010).
- Feng, Y., et al. A macrophage-activating, injectable hydrogel to sequester endogenous growth factors for in situ angiogenesis. Biomaterials. 134, 128-142 (2017).
- Lonez, C., et al. Cationic lipid nanocarriers activate Toll-like receptor 2 and NLRP3 inflammasome pathways. Nanomedicine: Nanotechnology, Biology, and Medicine. 10 (4), 775-782 (2014).
