

ERRATUM NOTICE
Important: There has been an erratum issued for this article. Read more …
Summary
Nous décrivons ici une méthode pour visualiser la synaptogenèse des neurones granulaires dans le cervelet de la souris au cours du développement cérébral postnatal lorsque ces cellules affinent leurs structures synaptiques et forment des synapses pour s’intégrer dans le circuit cérébral global.
Abstract
Les neurones subissent des changements dynamiques dans leur structure et leur fonction au cours du développement du cerveau pour former des connexions appropriées avec d’autres cellules. Le cervelet de rongeurs est un système idéal pour suivre le développement et la morphogenèse d’un seul type de cellule, le neurone granulaire cérébelleux (CGN), au fil du temps. Ici, l’électroporation in vivo des progéniteurs des neurones granulaires dans le cervelet de souris en développement a été utilisée pour marquer les cellules de manière clairsemée pour des analyses morphologiques ultérieures. L’efficacité de cette technique est démontrée dans sa capacité à mettre en évidence les étapes clés du développement de la maturation CGN, avec un accent particulier sur la formation de griffes dendritiques, qui sont des structures spécialisées où ces cellules reçoivent la majorité de leurs entrées synaptiques. En plus de fournir des instantanés des structures synaptiques CGN tout au long du développement cérébelleux, cette technique peut être adaptée pour manipuler génétiquement les neurones granulaires de manière autonome par les cellules afin d’étudier le rôle de tout gène d’intérêt et son effet sur la morphologie CGN, le développement des griffes et la synaptogenèse.
Introduction
Le développement du cerveau est un processus prolongé qui s’étend de l’embryogenèse à la vie postnatale. Pendant ce temps, le cerveau intègre une combinaison de stimuli intrinsèques et extrinsèques qui sculptent le câblage des synapses entre les dendrites et les axones pour finalement guider le comportement. Le cervelet des rongeurs est un système modèle idéal pour étudier le développement des synapses, car le développement d’un seul type de neurone, le neurone granulaire cérébelleux (CGN), peut être suivi lors de sa transition d’une cellule progénitrice à un neurone mature. Cela est dû, en partie, au fait qu’une majorité du cortex cérébelleux se développe après la naissance, ce qui permet une manipulation génétique facile et le marquage cellulaire après la naissance1.
Chez les mammifères, la différenciation CGN commence à la fin du développement embryonnaire lorsqu’un sous-ensemble de cellules prolifératives dans le cerveau postérieur migre sur la lèvre rhombique pour former une zone germinale secondaire à la surface du cervelet 2,3,4. Bien qu’elles soient pleinement engagées dans une identité de progéniteur de neurones granulaires (GNP), ces cellules continuent de proliférer dans la partie externe de la couche granulaire externe (EGL) jusqu’au jour postnatal 14 (P14). La prolifération de cette couche entraîne une expansion massive du cervelet car ces cellules donnent naissance exclusivement à CGNs5. Une fois que les CGN nouveau-nés quittent le cycle cellulaire dans l’EGL, ils migrent vers l’intérieur vers la couche granulaire interne (IGL), laissant derrière eux un axone qui bifurquera et voyagera dans la couche moléculaire du cervelet, formant des fibres parallèles qui synapsent sur les cellules de Purkinje6. La position de ces fibres dans la couche moléculaire dépend du moment de la sortie du cycle cellulaire.
Les CGN qui se différencient en premier quittent leurs fibres parallèles vers le bas de la couche moléculaire, tandis que les axones des CGN qui se différencient plus tard sont regroupés dans lehaut 7,8. Une fois que les corps cellulaires CGN atteignent l’IGL, ils commencent à élaborer des dendrites et à former des synapses avec des neurones inhibiteurs et excitateurs voisins. L’arbre dendritique mature d’une CGN présente une architecture stéréotypée avec quatre processus principaux. Au cours de la maturation CGN, les structures à l’extrémité de ces dendrites forment une griffe qui s’enrichit en protéines postsynaptiques 9,10. Ces structures spécialisées, appelées griffes dendritiques, contiennent la majorité des synapses sur les neurones granulaires et sont importantes pour recevoir à la fois les entrées excitatrices des innervations de fibres moussues provenant des pons, ainsi que les entrées inhibitrices des cellules de Golgi locales. Une fois entièrement configurées, les connexions synaptiques des CGN permettent à ces cellules de relayer les entrées des noyaux pré-cérébelleux aux cellules de Purkinje, qui se projettent hors du cortex cérébelleux vers les noyaux cérébelleux profonds.
L’électroporation postnatale in vivo des GNP est avantageuse par rapport à d’autres méthodes basées sur le marquage, telles que l’infection virale et la génération de lignées de souris transgéniques, car l’expression des constructions souhaitées peut être réalisée sur un calendrier rapide, et la méthode cible une petite population de cellules, utile pour étudier les effets autonomes cellulaires. Cette méthode a été utilisée dans des études antérieures pour étudier le développement morphologique des CGN; Cependant, ces études se sont concentrées sur un seul point temporel ou une courte fenêtre de temps 9,10,11,12,13. Cette méthode de marquage a été associée à une analyse d’images pour documenter les changements dans la morphologie CGN qui se produisent tout au long de la différenciation CGN au cours des trois premières semaines de la vie postnatale. Ces données révèlent la dynamique du développement des dendrites CGN qui sous-tend la construction des circuits cérébelleux.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
REMARQUE: Toutes les procédures ont été effectuées conformément aux protocoles approuvés par le Comité institutionnel de soin et d’utilisation des animaux de l’Université Duke (IACUC).
1. Préparation de l’ADN pour l’électroporation in vivo ou IVE (1 jour avant la chirurgie)
- Rassemblez les matériaux suivants : ADN purifié (0,5-25 μg par animal), acétate de sodium 3 M, éthanol, colorant vert rapide, eau distillée ultrapure, solution tampon phosphate (PBS) (voir le tableau des matériaux).
NOTE: Pour l’ADN, une construction exprimant la protéine fluorescente verte (GFP) sous un promoteur humain de l’ubiquitine a été obtenue à partir d’Addgene (FUGW, https://www.addgene.org/14883/). Toute construction exprimant la GFP ou une autre protéine fluorescente sous le contrôle d’un promoteur ubiquitaire devrait fonctionner. L’étiquetage spécifique à CGN avec cette technique ne dépend pas de la construction, mais plutôt de l’électroporation. - Préparer l’ADN pour l’électroporation en mélangeant la quantité désirée d’ADN, 10% en volume d’acétate de sodium 3 M et 250% en volume d’éthanol glacé à 100%. Notez que l’ADN précipitera immédiatement hors de la solution.
- Continuer à précipiter le mélange d’ADN pendant une nuit à -20 °C ou pendant une heure à -80 °C.
- La pastille précipitait l’ADN dans une centrifugeuse de table à 16 000 > × g et lavait deux fois avec de l’éthanol à 70%.
- Laisser sécher complètement la pastille d’ADN et reconstituer dans une solution 1x PBS + 0,02% Fast Green.

Figure 1 : Limitation de la profondeur d’injection à 1,5 mm à l’aide d’une entretoise. (A) Un segment de 11,2 mm est coupé d’une pipette de chargement à l’aide d’une lame de rasoir. (B) L’entretoise est fixée à l’extrémité de la seringue Hamilton (longueur totale de 1,27 cm ou 0,5 po) et fixée à l’aide d’adhésif ou de parafilm. La pointe exposée doit avoir une longueur de 1,5 mm. Veuillez cliquer ici pour voir une version agrandie de cette figure.
2. Électroporation in vivo de progéniteurs de neurones granulaires chez des souris postnatales âgées de sept jours
REMARQUE: Toutes les chirurgies d’électroporation ont été effectuées dans une salle d’opération stérile et hautement ventilée, et tout le personnel portait un équipement de protection individuelle complet, y compris des gants, un masque facial, un bonnet à cheveux, une blouse et des couvre-chaussures. Alternativement, les chirurgies peuvent être effectuées dans une hotte ventilée et stérile.
- Rassemblez le matériel suivant : ADN pour l’électroporation, petits ciseaux chirurgicaux, petites pinces chirurgicales, seringue Hamilton personnalisée, applicateur à embout de coton, coussin chauffant, bétadine, éthanol à 70 %, 1x PBS, parafilm, adhésif tissulaire (cyanoacrylate de n-butylester), isoflurane, électroporateur et électrodes de type pince à épiler (voir le tableau des matériaux).
- Couper une entretoise d’un embout de chargement stérilisé pour l’adapter à la seringue Hamilton afin de limiter la profondeur d’injection à 1,5 mm (figure 1A,B). Fixez l’entretoise avec de l’adhésif ou du parafilm.
- Anesthésier le chiot P7 dans une chambre d’isoflurane à un débit de 0,8 L/min. Confirmer l’anesthésie complète en surveillant la diminution de la respiration de l’animal et l’absence de pincement de l’orteil ou de la queue (figure 2A).
- Une fois que l’animal est complètement anesthésié, placez les petits sur un piédestal muni d’un cône nasal, délivrant constamment de l’isoflurane à 4 % à un débit de 0,8 L/min. Nettoyez le dessus de la tête du chiot 3 fois avec un tampon stérile de bétadine puis d’éthanol à 70%, en alternant entre les deux, pour préparer le site. Laisser sécher la solution avant de continuer.
- À l’aide d’une paire de ciseaux stérilisés, faites une petite incision avec une coupe qui s’étend du haut à la base des oreilles pour révéler le cerveau postérieur (figure 2B).
- Localisez le cervelet (figure 2C), insérez l’embout exposé de la seringue Hamilton dans le crâne, perpendiculairement au cerveau, et injectez 1,5 μL de mélange d’ADN dans le parenchyme cérébelleux en poussant lentement le piston arrière de la seringue. Après la livraison du mélange d’ADN, tirez lentement l’aiguille vers l’arrière pour éviter les déversements et laissez la solution d’ADN se diffuser pendant 30 s.
- Éteignez l’isoflurane et placez le chiot sur un coussin chauffant à 37 °C. Préparez l’électrode de type pince à épiler pour l’électroporation en plongeant les deux extrémités dans 1x PBS stérile.
REMARQUE: Le mouillage de l’électrode de type pince à épiler empêchera les brûlures de contact sur la peau du chiot pendant l’administration des impulsions électriques. - Orientez l’électrode de la pince à épiler au-dessus du site d’injection, l’extrémité plus étant orientée vers le bas et l’extrémité négative au-dessus de la tête de l’animal (figure 2D). Administrez cinq impulsions électriques de l’électroporateur avec les réglages suivants : intervalle d’impulsions de 50 ms, 130 V et 950 ms.
REMARQUE : Au besoin, effectuer une injection d’essai pour s’assurer que le site d’injection est situé sur le vermis cérébelleux (figure 2E). - Pincez l’incision fermée et scellez la plaie avec un adhésif tissulaire cyanoacrylate de n-butylester non toxique. Nettoyez la plaie avec de l’éthanol à 70%, car toute trace de sang augmente la probabilité d’infanticide parental et de cannibalisme.
- Laissez l’animal récupérer sur un coussin chauffant à 37 °C avant de le remettre au dirigé. Surveillez le(s) chiot(s) toutes les 30 minutes pendant au moins 2 heures après la chirurgie pour assurer un rétablissement complet.
NOTE: L’infanticide par l’un ou l’autre parent est assez courant. Pour éviter le cannibalisme, logez le père dans une cage différente avant de commencer l’électroporation et retournez toujours les chiots nettoyés et récupérés (c.-à-d. pas de tache de sang, complètement mobiles) dans la cage d’origine sur la litière d’origine. Les chiots peuvent également être essuyés avec les excréments de la cage d’origine pour minimiser l’odeur de sang. L’utilisation d’une mère porteuse peut être nécessaire si la mère d’origine continue de cannibaliser ses petits.

Figure 2 : Électroporation cérébelleuse in vivo de progéniteurs de neurones granulaires chez des bébés souris de type sauvage P7. (A) Les petits sont anesthésiés avec de l’isoflurane à 4% délivré à un taux de 0,8 L / min pour assurer l’anesthésie tout au long de l’injection de la solution d’ADN. L’isoflurane est délivré à un taux de 0,8 L/min. (B) Après avoir stérilisé la souris 3 fois avec de la bétadine et de l’éthanol à 70%, une incision est pratiquée qui s’étend sur la distance des oreilles, révélant le cerveau postérieur. (C) Image agrandie d’une démarcation blanche sur le crâne, point de repère pour le site d’injection. La construction de l’ADN doit être injectée à moins de 1 mm au-dessus de la marque; Des lignes pointillées délimitent la démarcation et une flèche noire indique le site d’injection. Les crêtes du vermis cérébelleux peuvent être visibles et peuvent être utiles pour trouver le site d’injection. (D) Orientation de l’électrode de type pince à épiler pour une électroporation efficace. L’extrémité plus (+) doit être orientée vers le bas pour attirer l’ADN chargé négativement dans le parenchyme cérébelleux avant l’administration d’impulsions électriques. (E) L’injection d’essai de 1 μL d’un colorant vert rapide à 0,02 % montre que l’injection est localisée au milieu du vermis cérébelleux entre les lobules 5 à 7. Veuillez cliquer ici pour voir une version agrandie de cette figure.
3. Immunohistochimie des CGN électroporés
- Rassemblez les matériaux suivants : isoflurane, 1x PBS, 4 % de paraformaldéhyde (PFA), 30 % de saccharose, sérum de chèvre normal, détergent non ionique, lames de verre, lames de verre, couvercles de verre, vernis à ongles, supports de montage, colorant nucléaire Hoechst et anticorps primaires et secondaires appropriés (voir le tableau des matériaux).
- Anesthésiez l’animal de laboratoire avec de l’isoflurane et confirmez l’anesthésie complète avec un pincement des orteils et de la queue.
- Effectuer une perfusion transcardique en injectant lentement 1x PBS et 4% PFA dans le ventricule gauche du cœur de l’animal. Laissez le sang s’écouler de l’animal en coupant la veine cave.
- Fixez le cerveau pendant la nuit en le submergeant dans 4% PFA à 4 ° C. Le lendemain, rincer rapidement le cerveau avec 1x PBS et transférer le cerveau dans 30% de saccharose dans 1x PBS pour la cryoprotection pendant au moins 24 heures.
- Si nécessaire, couper le cerveau en deux le long de l’axe rostral-caudale et confirmer l’expression de la construction du rapporteur transfecté à l’aide d’un microscope à dissection fluorescent vertical.
REMARQUE: Gardez le cerveau immergé dans 1x PBS dans un petit plat pour l’empêcher de se dessécher. - Montez le cerveau sur un microtome de congélation, coupez des sections sagittales de 25 μm et laissez les sections se dérouler dans un mélange 1:1 de 1x PBS et de glycérol.
NOTE: Les sections peuvent être stockées dans cette solution cryoprotectrice à -20 ° C pour un stockage à long terme. - Laver les sections trois fois dans 1x PBS pendant 10 min chacune pour éliminer le cryoprotecteur, et bloquer le tissu dans 1x PBS + 10% de sérum de chèvre normal + 0,2% de détergent non ionique sur un agitateur orbital à température ambiante pendant 1 h.
- Préparer la solution d’anticorps primaires: 1x PBS, 10% de sérum de chèvre normal, 0,2% de détergent non ionique et anticorps anti-GFP, et centrifuger la solution pendant 5 minutes à >16 000 × g. Incuber des sections dans la solution d’anticorps à 4 °C sur un agitateur orbital pendant 48 h.
- Laver la solution d’anticorps primaires pendant 15 min cinq fois avec 1x PBS + 0,2% de détergent non ionique.
- Préparer une solution d’anticorps secondaires : 1x PBS, 10 % de sérum de chèvre normal, 0,2 % de détergent non ionique et un anticorps secondaire approprié pour détecter la GFP; centrifuger la solution à 16 000 > × g. Incuber des sections dans la solution d’anticorps sur un agitateur orbital à température ambiante pendant 2-3 h. Protégez les sections de l’exposition à la lumière pour éviter le blanchiment.
- Laver la solution d’anticorps secondaires trois fois avec 1x PBS + 0,2% de détergent non ionique pendant 15 minutes à chaque fois. Incuber les sections dans 1x PBS + Hoechst pendant 5 min pour colorer les noyaux.
- Lavez la solution Hoechst avec 1x PBS + 0,2% de détergent non ionique et montez sur des lames de verre. Couvrez les sections avec un support de montage, recouvrez les lames et scellez la lame avec du vernis à ongles pour éviter l’évaporation.
4. Analyses morphologiques des CGN - reconstruction tridimensionnelle (3D) et surface et volume cellulaire
- Image de CGN électroporés simples sur un microscope confocal à objectif 63x avec un zoom 2x, prenant des images z-stack à 0,5 μm par pile. Imagez une cellule par fenêtre d’image pour faciliter l’analyse et la reconstruction de l’image.
- Installez le plug-in Simple Neurite Tracer pour FIJI en utilisant le lien suivant (https://imagej.net/Simple_Neurite_Tracer:_Basic_Instructions) pour tracer facilement et efficacement la structure des CGN électroporés dans l’espace tridimensionnel (3D).
Remarque : Il existe une version mise à jour du plug-in (https://imagej.net/SNT). - Analysez la longueur des neurites et la formation de griffes dendritiques en aveugle à l’aide de Simple Neurite Tracer. Téléchargez des images z-stack monocanal de CGN électroporées sur FIJI et cliquez sur Plugins | Segmentation | Traceur de neurite simple (Figure 3D).
- Accédez au menu déroulant et sélectionnez Créer une visionneuse 3D (Figure 3D).
- Faites défiler jusqu’à la base d’une dendrite, où elle se connecte à la cellule soma et commencez un chemin en cliquant sur la jonction. Tracez manuellement le chemin en cliquant sur les sections où le signal de remplissage de cellule est le plus lumineux, en appuyant sur [y] pour conserver la trace. Tracez jusqu’à l’extrémité de la dendrite si elle ne contient pas de griffe ou jusqu’à la base de la griffe et confirmez la trajectoire en appuyant sur [f] (Figure 4D).
- Ensuite, tracez la griffe en commençant un chemin à la base de la structure et en traçant jusqu’à la fin de la neurite la plus longue. Tracez les branches secondaires et tertiaires en maintenant [ctrl] enfoncé sous Windows ou [alt] sous Mac OS et en cliquant sur le chemin. Confirmez le chemin en appuyant sur [f].
- Observer que les mesures des traces sont visibles sur une fenêtre séparée; Additionnez toutes les mesures des branches de griffe (primaire, secondaire, tertiaire) pour obtenir la longueur totale de chaque griffe.
- Pour analyser la surface et le volume cellulaire des CGN électroporés, téléchargez le logiciel d’analyse cellulaire Imaris (https://imaris.oxinst.com/).
REMARQUE: FIJI peut également être utilisé pour reconstruire des cellules en 3D à partir d’images z-stack à l’aide de plug-ins facilement disponibles et gratuits. De plus, il existe une fonction de rendu volumétrique dans Simple Neurite Tracer, mais Imaris a été utilisé pour les raisons décrites ci-dessous. - Téléchargez l’image z-stack d’un CGN électroporé sur Imaris. Accédez à la boîte à outils de reconstruction 3D en appuyant sur Surpasser.
- Pour reconstruire le CGN, appuyez sur Surfaces et sélectionnez une région d’intérêt qui englobe l’intégralité de la cellule dans la fenêtre d’image. Une fois terminé, appuyez sur la flèche bleue vers l’avant dans le coin inférieur droit sous Créer.
- Si l’image contient plusieurs canaux pour différents signaux, sélectionnez le canal contenant le CGN électroporé et appuyez sur la flèche bleue vers l’avant.
- À l’aide du curseur, définissez le seuil souhaité qui correspond le plus précisément au signal de la cellule électroporée. Effectuez un zoom avant plus près de la surface de la cellule pour déterminer avec précision le seuil. Une fois terminé, appuyez sur la double flèche verte pour reconstruire la cellule et obtenir la surface et la taille du volume à partir des métadonnées.

Figure 3 : Analyse immunohistochimique et reconstruction tridimensionnelle de neurones granulaires électroporés. Des souris P7 CD-1 ont été électroporées avec une construction exprimant la GFP. Les cerveaux ont été collectés et soumis à l’immunohistochimie, à la microscopie confocale et à la reconstruction 3D pour l’analyse morphologique. (A) Chronologie entre l’électroporation et le traitement de l’image d’une souris à 10 DPI. B) Image de projection maximale d’une section transversale sagittale du cervelet électroporé 10-DPI; Les lignes blanches délimitent les couches cérébelleuses et la barre d’échelle est de 25 μm. (C) L’image de projection maximale d’un seul neurone granulaire électroporé 10-DPI et la trace 3D correspondante, la barre d’échelle est de 10 μm. (D) Les reconstructions 3D ont été générées à l’aide du plugin FIJI Simple Neurite Tracer. Toutes les mesures ont été tracées à travers la pile z, en suivant le signal de remplissage de cellule. Les mesures de l’arbre et des griffes ont été tracées séparément pour chaque dendrite; La ligne pointillée indique une partie de la dendrite dans le plan actuel. Abréviations : 3D = tridimensionnel; GFP = protéine fluorescente verte; DPI = jours après l’injection; PSD-95 = protéine de densité postsynaptique 95; PNB = progéniteurs de neurones granulaires; PFA = paraformaldéhyde. Veuillez cliquer ici pour voir une version agrandie de cette figure.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results

Figure 4 : Analyse de la morphologie des neurones granulaires au cours du développement cérébelleux. A) Images de projection maximale des CGN électroporés de 3 DPI à 14 DPI (âge postnatal P10 à P21), noyaux (bleu) et GFP (vert); Les pointes de flèches indiquent une dendrite individuelle et la barre d’échelle est de 10 μm. (B) Nombre moyen de dendrites. (C) Longueur moyenne de la dendrite mesurée de la base du soma à l’extrémité de la dendrite. (D) Fraction de dendrites qui contiennent une griffe; Une valeur de 1,00 est égale à 100%, c’est-à-dire que toutes les dendrites ont une griffe. (E) Longueur totale de la griffe dendritique. N > 30 cellules par affection, prélevées sur au moins 4 animaux par affection; toutes les mesures ont été analysées par ANOVA unidirectionnelle et soit un test de comparaison multiple de Dunnett (B, C et D) ou un test de comparaison multiple de Tukey (E), **** indique une signification avec p <0,0001 dans le temps; Les barres d’erreurs sont des abréviations S.E.M. : GFP = PROTÉINE FLUORESCENTE VERTE; DPI = jours après l’injection; PSD-95 = protéine de densité postsynaptique 95; CGN = neurones granulaires cérébelleux; ANOVA = analyse de la variance; S.E.M. = erreur-type de la moyenne. Veuillez cliquer ici pour voir une version agrandie de cette figure.
Pour étudier le développement de la morphologie des neurones granulaires in vivo, une construction exprimant la GFP sous le contrôle d’un promoteur humain de l’ubiquitine (FUGW) a été électroporée dans le cervelet en développement de souris CD-1 et de cerveaux prélevés 3, 5, 7, 10 et 14 jours après l’injection (DPI). Le marquage clairsemé des cellules par électroporation en combinaison avec la microscopie confocale capture des instantanés des CGN pendant les périodes d’élagage, de croissance et de maturation dendritiques. Pour analyser quantitativement et suivre la croissance des structures synaptiques CGN, chaque dendrite a été tracée à l’aide du plugin FIJI Simple Neurite Tracer (SNT). SNT est une méthode facile, rapide, efficace et facilement disponible pour mesurer la longueur des neurites et des griffes dans l’espace tridimensionnel (3D). Inversement, Imaris a été utilisé pour la reconstruction 3D des CGN afin d’obtenir des mesures de surface et de volume, car le programme fournit un rendu rapide et précis de chaque cellule, et ses capacités de seuillage sont capables d’isoler les cellules marquées des débris de cellules marquées à proximité.
Les CGN nouveau-nés subissent une phase exubérante de croissance dendritique suivie d’un raffinement de P10 à P14 (3 à 7-DPI) qui entraîne l’élagage de plus de 50% des dendrites en excès (Figure 4B). Cet événement coïncide avec l’allongement progressif des arbres restants (figure 4C) et la formation de structures en forme de griffes à l’extrémité de chaque dendrite (figure 4D), ce qui indique que ces processus de développement se produisent simultanément. Cependant, alors que les griffes sont trouvées sur environ 75% des dendrites par P14 (7-DPI), ces structures continuent d’augmenter en taille jusqu’à P21 (14-DPI) (Figure 4E).
Les changements dans la morphologie des dendrites et des griffes pourraient refléter soit un changement global de la taille totale des cellules, soit une redistribution de la membrane cellulaire. Pour répondre à cette question, chaque neurone granulaire marqué a été reconstruit dans Imaris pour quantifier la surface et le volume somatodendritiques totaux. La taille des CGN est restée relativement constante tout au long du développement (Figure 5A, B), bien qu’à P14, les CGN présentent une diminution significative de 20% du volume par rapport à P10, P12 et P17 (3-, 5- et 10-DPI) (Figure 5B). Ces données suggèrent que le recyclage membranaire des dendrites rétractées peut être particulièrement important pour permettre l’élargissement des terminaisons dendritiques en griffes et indiquent que P14 (7-DPI) est un moment clé dans la transition de l’élagage au développement des synapses.

Figure 5 : Analyse de la taille des neurones granulaires au cours du développement cérébelleux. Les CGN électroporés ont été reconstruits à Imaris pour déterminer la taille cellulaire. (A-B) Analyse de surface et volumétrique des neurones granulaires (c.-à-d. soma cellulaire et dendrites) au cours du développement cérébelleux. N > 30 cellules par affection, prélevées sur au moins 4 animaux par affection. Toutes les mesures ont été analysées par ANOVA unidirectionnelle et un test de comparaison multiple de Dunnett, ** indique une signification avec p <0,005; Les barres d’erreurs sont des abréviations S.E.M. : DPI = JOURS APRÈS L’INJECTION; CGN = neurones granulaires cérébelleux; ANOVA = analyse de la variance; S.E.M. = erreur-type de la moyenne. Veuillez cliquer ici pour voir une version agrandie de cette figure.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Les neurones granulaires cérébelleux sont les neurones les plus abondants dans le cerveau des mammifères, représentant près de 60 à 70% de la population totale de neurones dans le cerveau des rongeurs 1,14. Le cervelet a été largement utilisé pour élucider les mécanismes de prolifération cellulaire, de migration, de formation de dendrites et de développement des synapses 6,9,10,11,15,16,17,18,19,20 . De plus, des décennies d’études électrophysiologiques ont permis d’établir le rôle des neurones granulaires dans la plasticité cérébelleuse à long terme, qui sous-tend l’implication du circuit dans les comportements moteurs appris21,22,23. Ainsi, les neurones granulaires sont un excellent système modèle pour interroger les questions clés concernant le développement des synapses et des circuits.
Le but de cette étude est de mettre en évidence une technique qui tire parti du cervelet pour suivre la morphologie d’un seul type de neurone in vivo. L’électroporation du cervelet cible les cellules en division; Ainsi, il est techniquement capable de marquer à la fois les progéniteurs des neurones granulaires et les cellules gliales, bien que parce que les progéniteurs des neurones granulaires sont si nombreux, la population électroporée est en grande partie neuronale, même sans avoir besoin de promoteurs spécifiques au type cellulaire. De plus, cette technique peut être adaptée pour manipuler génétiquement des gènes in vivo afin d’étudier leurs rôles dans le développement de CGN. Ceci peut être réalisé par transfection d’un plasmide exprimant soit des ARN courts en épingle à cheveux ou de petits ARN interférents pour abattre des gènes ou un plasmide exprimant la Cre recombinase en une souche de souris transgénique pour exciser les régions flanquées de sites LoxP et éliminer le gène d’intérêtprévu 10,24.
L’électroporation a à la fois des forces et des faiblesses pour les études génétiques. Il offre une méthode plus rapide pour manipuler les gènes par rapport aux stratégies transgéniques traditionnelles, bien que la rareté de l’électroporation limite son utilité pour les études comportementales, qui nécessitent un grand nombre de cellules génétiquement modifiées pour voir les effets. Une deuxième limitation est qu’il ne cible de manière fiable que les neurones granulaires entre les lobules 5 et 7, et cela est dû, en partie, à la façon dont le cervelet est orienté au site d’injection. Cependant, dans la région électroporée, cette méthode produit un nombre relativement important de cellules marquées individuellement, ce qui permet des mesures statistiquement robustes. Par exemple, non seulement il a été possible de suivre le développement des dendrites au fil du temps, mais aussi des redistributions subtiles de la membrane entre les dendrites et les griffes ont pu être mesurées, en étendant les données précédemment publiées20. Les griffes CGN sont particulièrement intéressantes à étudier car l’élargissement de ces structures dendritiques fournit un espace supplémentaire aux neurones granulaires pour former à la fois des connexions excitatrices avec des bornes de fibres moussues et des cellules de brosse unipolaires ainsi que des connexions inhibitrices avec les cellules de Golgi voisines. Par conséquent, la combinaison de ces mesures dendritiques avec le marquage immunohistochimique de protéines pré- et postsynaptiques spécifiques pourrait être utile pour faire progresser l’étude de la formation du circuit cérébelleux et de la maturation des synapses.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Les auteurs ne déclarent aucun conflit d’intérêts.
Acknowledgments
Le travail a été soutenu par les subventions NIH R01NS098804 (A.E.W.), F31NS113394 (U.C.) et le programme d’été en neurosciences de l’Université Duke (D.G.).
Materials
| Name | Company | Catalog Number | Comments |
| Betadine | Purdue Production | 67618-150-17 | |
| Cemented 10 µL needle | Hamilton | 1701SN (80008) | 33 gauge, 1.27 cm (0.5 in), 4 point style |
| Chicken anti-GFP | Millipore Sigma | AB16901 | Our lab uses this antibody at a 1:1000 concentration |
| Cotton-tip applicator | |||
| Donkey anti-chicken Cy2 | Jackson ImmunoResearch | 703-225-155 | Our lab uses this antibody at a 1:500 concentration |
| Ethanol (200 proof) | Koptec | V1016 | |
| Electroporator ECM 830 | BTX Harvard Apparatus | 45-0052 | |
| Fast Green FCF | Sigma | F7252-5G | |
| FUGW plasmid | Addgene | 14883 | |
| Glass slides | VWR | 48311-703 | Superfrost plus |
| Glycerol | Sigma-Aldrich | G5516 | |
| Heating pad | Softheat | ||
| Hoescht 33342 fluorescent dye | Invitrogen | 62249 | |
| Imaris | Bitplane | ||
| Isoflurane | Patterson Veterinary | 07-893-1389 | |
| Micro cover glass | VWR | 48382-138 | |
| Nail polish | Sally Hansen | Color 109 | |
| Normal goat serum | Gibco | 16210064 | |
| O.C.T. embedding compound | Tissue-Tek | 4583 | |
| Olympus MVX10 Dissecting Scope | Olympus | MVX10 | |
| P200 pipette reach tip | Fisherbrand | 02-707-138 | Used for needle spacer |
| Parafilm | Bemis | PM-996 | |
| PBS pH 7.4 (10x) | Gibco | 70011-044 | |
| Simple Neurite Tracer | FIJI | https://imagej.net/Simple_Neurite_Tracer:_Basic_ Instructions |
|
| Sucrose | Sigma | S0389 | |
| Surgical tools | RWD Life Science | Small scissors and tweezers | |
| Triton X-100 | Roche | 11332481001 | non-ionic detergent |
| Tweezertrodes | BTX Harvard Apparatus | 45-0489 | 5 mm, platinum plated tweezer-type electrodes |
| Ultrapure distilled water | Invitrogen | 10977-015 | |
| Vectashield mounting media | Vectashield | H1000 | |
| Vetbond tissue adhesive | 3M | 1469SB | |
| Zeiss 780 Upright Confocal | Zeiss | 780 |
References
- Altman, J., Bayer, S. A. Development of the cerebellar system : in relation to its evolution, structure, and functions. , CRC Press. (1997).
- Rahimi-Balaei, M., Bergen, H., Kong, J., Marzban, H. Neuronal migration during development of the cerebellum. Frontiers in Cellular Neuroscience. 12, 484 (2018).
- Alder, J., Cho, N. K., Hatten, M. E. Embryonic precursor cells from the rhombic lip are specified to a cerebellar granule neuron identity. Neuron. 17 (3), 389-399 (1996).
- Hatten, M. E., Heintz, N. Mechanisms of neural patterning and specification in the developing cerebellum. Annual Review of Neuroscience. 18, 385-408 (1995).
- Ben-Arie, N., et al. Math1 is essential for genesis of cerebellar granule neurons. Nature. 390 (6656), 169-172 (1997).
- Borghesani, P. R., et al. BDNF stimulates migration of cerebellar granule cells. Development. 129 (6), 1435-1442 (2002).
- Espinosa, J. S., Luo, L. Timing neurogenesis and differentiation: insights from quantitative clonal analyses of cerebellar granule cells. Journal of Neuroscience. 28 (10), 2301-2312 (2008).
- Markwalter, K. H., Yang, Y., Holy, T. E., Bonni, A. Sensorimotor coding of vermal granule neurons in the developing mammalian cerebellum. Journal of Neuroscience. 39 (34), 6626-6643 (2019).
- Shalizi, A., et al. PIASx is a MEF2 SUMO E3 ligase that promotes postsynaptic dendritic morphogenesis. Journal of Neuroscience. 27 (37), 10037-10046 (2007).
- Shalizi, A., et al. A Calcium-regulated MEF2 sumoylation switch controls poststynaptic differentiation. Science. 311 (5763), 1012-1017 (2006).
- Konishi, Y., Stegmuller, J., Matsuda, T., Bonni, S., Bonni, A. Cdh1-APC controls axonal growth and patterning in the mammalian brain. Science. 303 (5660), 1026-1030 (2004).
- Holubowska, A., Mukherjee, C., Vadhvani, M., Stegmuller, J. Genetic manipulation of cerebellar granule neurons in vitro and in vivo to study neuronal morphology and migration. Journal of Visualized Experiments: JoVE. (85), e51070 (2014).
- Yang, Y., et al. Chromatin remodeling inactivates activity genes and regulates neural coding. Science. 353 (6296), 300-305 (2016).
- Herculano-Houzel, S. Coordinated scaling of cortical and cerebellar numbers of neurons. Frontiers in Neuroanatomy. 4, 12 (2010).
- Wilson, P. M., Fryer, R. H., Fang, Y., Hatten, M. E. Astn2, a novel member of the astrotactin gene family, regulates the trafficking of ASTN1 during glial-guided neuronal migration. Journal of Neuroscience. 30 (25), 8529-8540 (2010).
- Kokubo, M., et al. BDNF-mediated cerebellar granule cell development is impaired in mice null for CaMKK2 or CaMKIV. Journal of Neuroscience. 29 (28), 8901-8913 (2009).
- Schwartz, P. M., Borghesani, P. R., Levy, R. L., Pomeroy, S. L., Segal, R. A. Abnormal cerebellar development and foliation in BDNF-/- mice reveals a role for neurotrophins in CNS patterning. Neuron. 19 (2), 269-281 (1997).
- Segal, R. A., Pomeroy, S. L., Stiles, C. D. Axonal growth and fasciculation linked to differential expression of BDNF and NT3 receptors in developing cerebellar granule cells. Journal of Neuroscience. 15 (7), Pt 1 4970-4981 (1995).
- Zhou, P., et al. Polarized signaling endosomes coordinate BDNF-induced chemotaxis of cerebellar precursors. Neuron. 55 (1), 53-68 (2007).
- Dhar, M., Hantman, A. W., Nishiyama, H. Developmental pattern and structural factors of dendritic survival in cerebellar granule cells in vivo. Scientific Reports. 8 (1), 17561 (2018).
- Ito, M. Synaptic plasticity in the cerebellar cortex and its role in motor learning. Canadian Journal of Neurological Sciences. 20, Suppl 3 70-74 (1993).
- Jorntell, H., Hansel, C. Synaptic memories upside down: bidirectional plasticity at cerebellar parallel fiber-Purkinje cell synapses. Neuron. 52 (2), 227-238 (2006).
- Nakanishi, S. Genetic manipulation study of information processing in the cerebellum. Neuroscience. 162 (3), 723-731 (2009).
- Chang, C. H., et al. Atoh1 controls primary cilia formation to allow for SHH-triggered granule neuron progenitor proliferation. Developmental Cell. 48 (2), 184-199 (2019).
Tags
Neurosciences numéro 172 Développement neuronal cervelet dendrite synapse neurone granulaire électroporationErratum
Formal Correction: Erratum: Utilizing In Vivo Postnatal Electroporation to Study Cerebellar Granule Neuron Morphology and Synapse Development
Posted by JoVE Editors on 04/06/2023.
Citeable Link.
An erratum was issued for: Utilizing In Vivo Postnatal Electroporation to Study Cerebellar Granule Neuron Morphology and Synapse Development. A figure was updated.
Figure 2 was updated from:
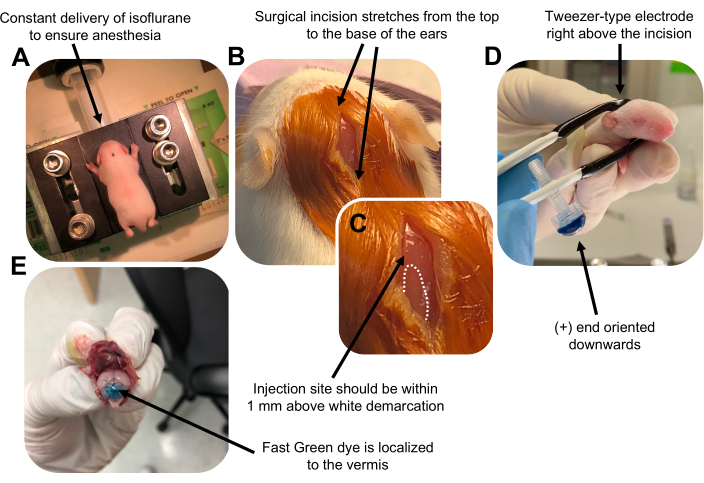
Figure 2: In vivo cerebellar electroporation of granule neuron progenitors in P7 wildtype mouse pups. (A) Pups are anesthetized with 4% isoflurane delivered at a rate of 0.8L/min to ensure anesthesia throughout the injection of the DNA solution. Isoflurane is delivered at a rate of 0.8 L/min. (B) After sterilizing the mouse 3 times with betadine and 70% ethanol, an incision is made that spans the distance of the ears, revealing the hindbrain. (C) A magnified image of a white demarcation on the cranium, a landmark for the injection site. DNA construct should be injected within 1 mm above the mark; dotted lines outline the demarcation, and black arrow denotes the injection site. The ridges of the cerebellar vermis may be visible and can be useful for finding the injection site. (D) Tweezer-type electrode orientation for efficient electroporation. Plus (+) end must be oriented downwards to pull negatively charged DNA into the cerebellar parenchyma prior to administration of electrical pulses. (E) Test injection of 1 µL of a 0.02% Fast Green dye shows injection is localized to the middle of the cerebellar vermis between lobules 5-7. Please click here to view a larger version of this figure.
to:
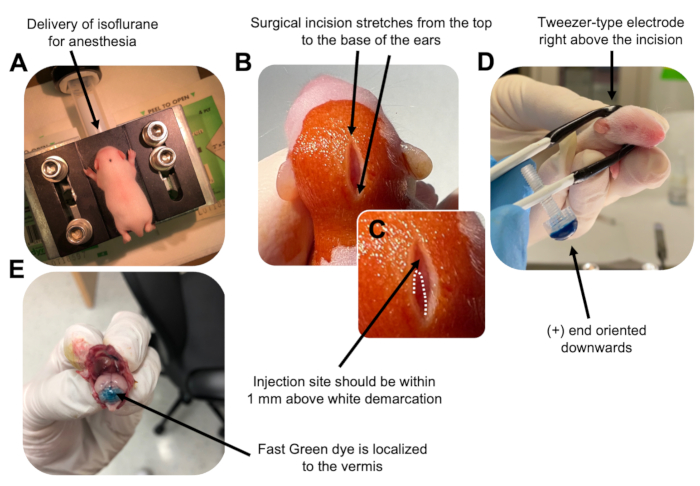
Figure 2: In vivo cerebellar electroporation of granule neuron progenitors in P7 wildtype mouse pups. (A) Pups are anesthetized with 4% isoflurane delivered at a rate of 0.8L/min to ensure anesthesia throughout the injection of the DNA solution. Isoflurane is delivered at a rate of 0.8 L/min. (B) After sterilizing the mouse 3 times with betadine and 70% ethanol, an incision is made that spans the distance of the ears, revealing the hindbrain. (C) A magnified image of a white demarcation on the cranium, a landmark for the injection site. DNA construct should be injected within 1 mm above the mark; dotted lines outline the demarcation, and black arrow denotes the injection site. The ridges of the cerebellar vermis may be visible and can be useful for finding the injection site. (D) Tweezer-type electrode orientation for efficient electroporation. Plus (+) end must be oriented downwards to pull negatively charged DNA into the cerebellar parenchyma prior to administration of electrical pulses. (E) Test injection of 1 µL of a 0.02% Fast Green dye shows injection is localized to the middle of the cerebellar vermis between lobules 5-7. Please click here to view a larger version of this figure.
