NOTE: All procedures were performed under protocols approved by Duke University Institutional Animal Care and Use Committee (IACUC).
1. DNA preparation for in vivo electroporation or IVE (1 day before surgery)
- Gather the following materials: purified DNA (0.5-25 µg per animal), 3 M sodium acetate, ethanol, Fast Green dye, ultrapure distilled water, phosphate buffer solution (PBS) (see the Table of Materials).
NOTE: For DNA, a construct expressing green fluorescent protein (GFP) under a human ubiquitin promoter was obtained from Addgene (FUGW, https://www.addgene.org/14883/). Any construct expressing GFP or another fluorescent protein under the control of a ubiquitous promoter should work. CGN-specific labeling with this technique is not dependent on the construct, but rather on the electroporation. - Prepare DNA for electroporation by mixing the desired amount of DNA, 10% by volume of 3 M sodium acetate and 250% by volume of 100% ice-cold ethanol. Note that the DNA will precipitate out of solution immediately.
- Continue to precipitate the DNA mixture overnight at -20 °C or for an hour at -80 °C.
- Pellet precipitated DNA in a tabletop centrifuge at >16,000 × g and wash twice with 70% ethanol.
- Allow the DNA pellet to dry completely, and reconstitute in a 1x PBS + 0.02% Fast Green solution.

Figure 1: Limiting injection depth to 1.5 mm using a spacer. (A) A 11.2 mm segment is cut off a loading pipette using a razor blade. (B) The spacer is fitted onto the tip of the Hamilton syringe (total length is 1.27 cm or 0.5 in) and secured with either adhesive or parafilm. The exposed tip should be 1.5 mm in length. Please click here to view a larger version of this figure.
2. In vivo electroporation of granule neuron progenitors in seven-day old postnatal mice
NOTE: All electroporation surgeries were performed in a sterile and highly ventilated surgical suite, and all personnel wore complete personal protective equipment including gloves, face mask, hair bonnet, gown, and shoe covers. Alternatively, the surgeries can be performed in a ventilated and sterile hood.
- Gather the following materials: DNA for electroporation, small surgical scissors, small surgical tweezers, customized Hamilton syringe, cotton-tip applicator, heating pad, betadine, 70% ethanol, 1x PBS, parafilm, tissue adhesive (n-butyl-ester cyanoacrylate), isoflurane, electroporator, and tweezer-type electrodes (see the Table of Materials).
- Cut a spacer from a sterilized loading tip to fit over the Hamilton syringe to limit the injection depth to 1.5 mm (Figure 1A,B). Secure the spacer with adhesive or parafilm.
- Anesthetize the P7 pup in an isoflurane chamber at a delivery rate of 0.8 L/min. Confirm full anesthesia by monitoring the animal for decreased respiration and a lack of a toe or tail pinch response (Figure 2A).
- Once the animal is fully anaesthetized, place the pups on a pedestal fitted with a nose cone, delivering constant 4% isoflurane at a delivery rate of 0.8 L/min. Clean the top of the pup's head 3 times with a sterile swab of betadine then 70% ethanol, alternating between the two, to prepare the site. Allow the solution to dry before proceeding.
- Using a pair of sterilized scissors, make a small incision with one cut that spans the distance from the top to the base of the ears to reveal the hindbrain (Figure 2B).
- Locate the cerebellum (Figure 2C), insert the exposed tip of the Hamilton syringe through the skull, perpendicular to the brain, and inject 1.5 µL of DNA mixture into the cerebellar parenchyma by slowly pushing the back plunger of the syringe. After delivery of the DNA mixture, slowly pull the needle back to prevent back spills, and allow the DNA solution to diffuse for 30 s.
- Turn off the isoflurane, and place the pup on a 37 °C heating pad. Prepare the tweezer-type electrode for electroporation by dipping both ends into sterile 1x PBS.
NOTE: Wetting the tweezer-type electrode will prevent contact burns on the skin of the pup during administration of the electrical pulses. - Orient the tweezer-electrode above the site of injection with the plus end facing downwards and the negative end above the animal's head (Figure 2D). Administer five electrical pulses from the electroporator with the following settings: 50 ms, 130 V, and 950 ms inter-pulse interval.
NOTE: If needed, perform a test injection to ensure that the injection site is located on the cerebellar vermis (Figure 2E). - Pinch the incision closed, and seal the wound with a non-toxic n-butyl-ester cyanoacrylate tissue adhesive. Clean the wound with 70% ethanol as any trace amount of blood increases the likelihood of parental infanticide and cannibalism.
- Allow the animal to recover on a 37 °C heating pad before returning the pup to the dam. Monitor the pup(s) every 30 min for at least 2 h after the surgery to ensure full recovery.
NOTE: Infanticide by either parent is quite common. To prevent cannibalism, house the sire in a different cage before starting the electroporation, and always return cleaned and recovered pups (i.e., no bloodstain, fully mobile) to the original cage on the original bedding. Pups can also be wiped with droppings from the original cage to minimize the smell of blood. The use of a surrogate dam may be necessary if the original dam continues to cannibalize her pups.
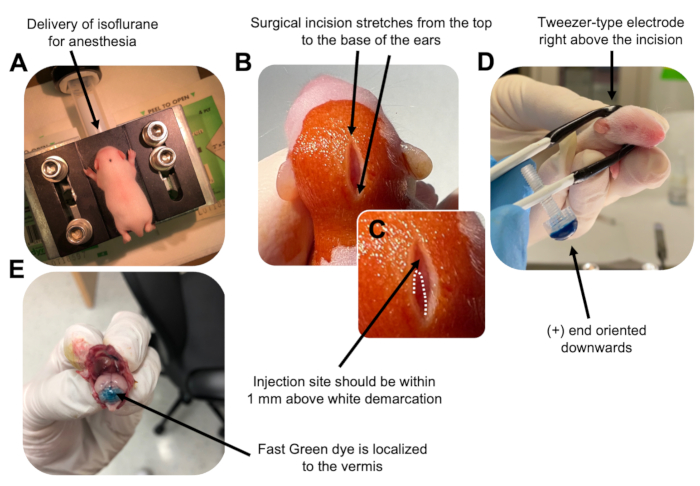
Figure 2: In vivo cerebellar electroporation of granule neuron progenitors in P7 wildtype mouse pups. (A) Pups are anesthetized with 4% isoflurane delivered at a rate of 0.8L/min to ensure anesthesia throughout the injection of the DNA solution. Isoflurane is delivered at a rate of 0.8 L/min. (B) After sterilizing the mouse 3 times with betadine and 70% ethanol, an incision is made that spans the distance of the ears, revealing the hindbrain. (C) A magnified image of a white demarcation on the cranium, a landmark for the injection site. DNA construct should be injected within 1 mm above the mark; dotted lines outline the demarcation, and black arrow denotes the injection site. The ridges of the cerebellar vermis may be visible and can be useful for finding the injection site. (D) Tweezer-type electrode orientation for efficient electroporation. Plus (+) end must be oriented downwards to pull negatively charged DNA into the cerebellar parenchyma prior to administration of electrical pulses. (E) Test injection of 1 µL of a 0.02% Fast Green dye shows injection is localized to the middle of the cerebellar vermis between lobules 5-7. Please click here to view a larger version of this figure.
3. Immunohistochemistry of electroporated CGNs
- Gather the following materials: isoflurane, 1x PBS, 4% paraformaldehyde (PFA), 30% sucrose, normal goat serum, non-ionic detergent, glass slides, glass coverslips, nail polish, mounting media, Hoechst nuclear dye, and appropriate primary and secondary antibodies (see the Table of Materials).
- Anesthetize the experimental animal with isoflurane, and confirm full anesthesia with a toe and tail pinch.
- Perform a trans-cardial perfusion by slowly injecting 1x PBS and 4% PFA into the left ventricle of the animal's heart. Allow the blood to drain from the animal by cutting the vena cava.
- Fix the brain overnight by submerging it in 4% PFA at 4 °C. On the following day, quickly rinse the brain with 1x PBS, and transfer the brain into 30% sucrose in 1x PBS for cryoprotection for at least 24 h.
- If necessary, slice the brain in half along the rostral-caudal axis, and confirm expression of the transfected reporter construct using an upright fluorescent dissecting microscope.
NOTE: Keep the brain submerged in 1x PBS in a small dish to prevent it from drying out. - Mount the brain on a freezing microtome, slice 25 µm sagittal sections, and allow sections to unfold in a 1:1 mixture of 1x PBS and glycerol.
NOTE: Sections can be stored in this cryoprotectant solution at -20 °C for long-term storage. - Wash sections three times in 1x PBS for 10 min each to remove cryoprotectant, and block the tissue in 1x PBS + 10% normal goat serum + 0.2% non-ionic detergent on an orbital shaker at room temperature for 1 h.
- Prepare primary antibody solution: 1x PBS, 10% normal goat serum, 0.2% non-ionic detergent, and anti-GFP antibody, and centrifuge the solution for 5 min at >16,000 × g. Incubate sections in the antibody solution at 4 °C on an orbital shaker for 48 h.
- Wash off primary antibody solution for 15 min five times with 1x PBS + 0.2% non-ionic detergent.
- Prepare secondary antibody solution: 1x PBS, 10% normal goat serum, 0.2% non-ionic detergent, and an appropriate secondary antibody to detect GFP; centrifuge the solution at >16,000 × g. Incubate sections in the antibody solution on an orbital shaker at room temperature for 2-3 h. Protect the sections from light exposure to prevent bleaching.
- Wash off secondary antibody solution three times with 1x PBS + 0.2% non-ionic detergent for 15 min each time. Incubate sections in 1x PBS + Hoechst for 5 min to stain nuclei.
- Wash off Hoechst solution with 1x PBS + 0.2% non-ionic detergent and mount onto glass slides. Cover the sections with mounting media, coverslip the slides, and seal the slide with nail polish to prevent evaporation.
4. Morphological analyses of CGNs – three-dimensional (3D) reconstruction and surface area and cellular volume
- Image single electroporated CGNs on a confocal microscope at 63x objective with a 2x zoom, taking z-stack images at 0.5 µm per stack. Image one cell per image window to allow for easy image analysis and reconstruction.
- Install the Simple Neurite Tracer plug-in for FIJI using the following link (https://imagej.net/Simple_Neurite_Tracer:_Basic_Instructions) to easily and efficiently trace the structure of electroporated CGNs in three-dimensional (3D) space.
NOTE: There is an updated version of the plug-in (https://imagej.net/SNT). - Analyze neurite length and dendritic claw formation in a blinded manner using Simple Neurite Tracer. Upload single-channel z-stack images of electroporated CGNs onto FIJI, and click on Plugins | Segmentation | Simple Neurite Tracer (Figure 3D).
- Access the drop-down menu, and select Create New 3D Viewer (Figure 3D).
- Scroll to the base of a dendrite, where it connects to the cell soma and start a path by clicking on the junction. Manually trace the path by clicking through the sections where the cell-fill signal is brightest, pressing [y] to keep the trace. Trace until the end of the dendrite if it does not contain a claw or until the base of the claw and confirm the path by pressing [f] (Figure 4D).
- Next, trace the claw by starting a path at the base of the structure and tracing until the end of the longest neurite. Trace secondary and tertiary branches by holding down [ctrl] on Windows or [alt] on a Mac OS and clicking the path. Confirm the path by pressing [f].
- Observe that measurements for the traces are visible on a separate window; add up all of the measurements of the claw branches (primary, secondary, tertiary) to obtain the total length for each claw.
- For analyzing surface area and cellular volume of electroporated CGNs, download Imaris cell analysis software (https://imaris.oxinst.com/).
NOTE: FIJI can also be used to reconstruct cells in 3D from z-stack images using readily available and free plug-ins. Additionally, there is a volumetric rendering feature in Simple Neurite Tracer but Imaris was used for the reasons outlined below. - Upload z-stack image of an electroporated CGN onto Imaris. Access the 3D-reconstruction toolkit by pressing Surpass.
- To reconstruct the CGN, press Surfaces, and select a region of interest that encompasses the entirety of the cell within the image window. Once finished, press the blue forward arrow at the bottom right corner under Create.
- If the image contains multiple channels for different signals, select the channel containing the electroporated CGN, and press the blue forward arrow.
- Using the slidebar, set a desired threshold that most accurately fits the signal of the electroporated cell. Zoom in closer to the surface of the cell to accurately determine the threshold. Once finished, press the double green arrow to reconstruct the cell and obtain the surface area and volume size from the metadata.

Figure 3: Immunohistochemical analysis and three-dimensional reconstruction of electroporated granule neurons. P7 CD-1 mice were electroporated with a construct expressing GFP. Brains were collected and subjected to immunohistochemistry, confocal microscopy, and 3D-reconstruction for morphological analysis. (A) Timeline from electroporation to image processing of a 10-DPI mouse. (B) Maximum projection image of a sagittal cross-section of electroporated cerebellum 10-DPI; white lines demarcate cerebellar layers, and scale bar is 25 µm. (C) Maximum projection image of a single electroporated granule neuron 10-DPI and the corresponding 3D trace, scale bar is 10 µm. (D) 3D-reconstructions were generated using the FIJI plugin Simple Neurite Tracer. All measurements were traced through the z-stack, following the cell-fill signal. Shaft and claw measurements were traced separately for every dendrite; dotted line denotes portion of dendrite within the current plane. Abbreviations: 3D = three-dimensional; GFP = green fluorescent protein; DPI = days post-injection; PSD-95 = postsynaptic density protein 95; GNPs = granule neuron progenitors; PFA = paraformaldehyde. Please click here to view a larger version of this figure.

Figure 4: Analysis of granule neuron morphology during cerebellar development. (A) Maximum projection images of electroporated CGNs from 3-DPI to 14-DPI (postnatal age P10 to P21), nuclei (blue) and GFP (green); arrowheads indicate individual dendrite, and scale bar is 10 µm. (B) Average number of dendrites. (C) Average dendrite length measured from the base of the soma to the tip of the dendrite. (D) Fraction of dendrites that contain a claw; a value of 1.00 is 100%, i.e., all dendrites have claw. (E) Total length of dendritic claw. N > 30 cells per condition, collected from at least 4 animals per condition; all measurements were analyzed by one-way ANOVA and either a Dunnett's multiple comparison test (B, C, and D) or a Tukey's multiple comparison test (E), **** denotes significance with p <0.0001 across time; errors bars are S.E.M. Abbreviations: GFP = green fluorescent protein; DPI = days post-injection; PSD-95 = postsynaptic density protein 95; CGNs = cerebellar granule neurons; ANOVA = analysis of variance; S.E.M. = standard error of the mean. Please click here to view a larger version of this figure.
To study the development of granule neuron morphology in vivo, a construct expressing GFP under the control of a human ubiquitin promoter (FUGW) was electroporated into the developing cerebellum of CD-1 mice and brains collected 3-, 5-, 7-, 10-, and 14-days post-injection (DPI). Sparse labeling of cells by electroporation in combination with confocal microscopy capture snapshots of CGNs during periods of dendritic pruning, growth, and maturation. To quantitatively analyze and track the growth of CGN synaptic structures, each dendrite was traced using the FIJI plugin Simple Neurite Tracer (SNT). SNT is an easy, fast, efficient, and readily available method to measure neurite and claw length in three-dimensional (3D) space. Conversely, Imaris was used for 3D-reconstruction of CGNs to obtain surface area and volume measurements because the program provides fast and accurate rendering of each cell, and its thresholding capabilities are able to isolate labeled cells from nearby labeled cell debris.
Newborn CGNs undergo an exuberant phase of dendritic growth followed by refinement from P10 to P14 (3- to 7-DPI) that results in the pruning of more than 50% of excess dendrites (Figure 4B). This event coincides with the gradual lengthening of the remaining arbors (Figure 4C) and the formation of claw-like structures at the end of each dendrite (Figure 4D), indicating that these developmental processes are happening concurrently. However, while claws are found on roughly 75% of dendrites by P14 (7-DPI), these structures continue to increase in size until P21 (14-DPI) (Figure 4E).
The changes in dendrite and claw morphology could reflect either an overall change in total cell size or a redistribution of cell membrane. To address this question, each labeled granule neuron was reconstructed in Imaris to quantify the total somatodendritic surface area and volume. CGN size remained relatively constant across development (Figure 5A,B), though at P14, CGNs exhibit a significant 20% decrease in volume compared to P10, P12, and P17 (3-, 5-, and 10-DPI) (Figure 5B). These data suggest that membrane recycling from retracted dendrites may be particularly important for permitting the enlargement of dendritic endings into claws and point to P14 (7-DPI) as a key time point in the transition from pruning to synapse development.

Figure 5: Analysis of granule neuron size during cerebellar development. Electroporated CGNs were reconstructed in Imaris to determine cellular size. (A–B) Surface area and volumetric analysis of granule neurons (i.e., cell soma and dendrites) during cerebellar development. N > 30 cells per condition, collected from at least 4 animals per condition. All measurements were analyzed by one-way ANOVA and a Dunnett's multiple comparison test, ** denotes significance with p <0.005; errors bars are S.E.M. Abbreviations: DPI = days post-injection; CGNs = cerebellar granule neurons; ANOVA = analysis of variance; S.E.M. = standard error of the mean. Please click here to view a larger version of this figure.
