



Overview
Fonte: Alexander S. Gold1, Tonya M. Colpitts1
1 Dipartimento di Microbiologia, Boston University School of Medicine, National Emerging Infections Diseases Laboratories, Boston, MA
La trasduzione è una forma di scambio genetico tra batteri che utilizza batteriofagi, o fagi, una classe di virus che infetta esclusivamente organismi procariotici. Questa forma di trasferimento del DNA, da un batterio all'altro attraverso un fago, fu scoperta nel 1951 da Norton Zinder e Joshua Ledererg, che definirono il processo "trasduzione" (1). I batteriofagi furono scoperti per la prima volta nel 1915 dal batteriologo britannico Frederick Twort, poi scoperti indipendentemente nel 1917 dal microbiologo franco-canadese Felix d'Herelle (2). Da allora, la struttura e la funzione di questi fagi sono state ampiamente caratterizzate (3), dividendo questi fagi in due classi. La prima di queste classi sono i fagi litici che al momento dell'infezione si moltiplicano all'interno del batterio ospite, interrompendo il metabolismo batterico, lisi della cellula e rilasciando fagi di progenie (4). Come risultato di questa attività antibatterica e della crescente prevalenza di batteri resistenti agli antibiotici, questi fagi litici si sono recentemente dimostrati utili come trattamento sostitutivo degli antibiotici. La seconda di queste classi sono i fagi lisogenici che possono moltiplicarsi all'interno dell'ospite attraverso il ciclo litico o entrare in uno stato quiescente in cui il loro genoma è integrato in quello dell'ospite (Figura 1), un processo noto come lisogenesi, con la capacità di indotta dalla produzione di fagi in più generazioni successive (4).
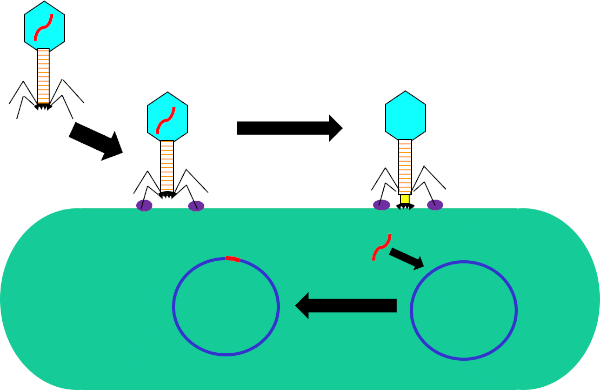
Figura 1: Infezione della cellula ospite da parte del batteriofago. Adsorbimento da parte del fago alla parete cellulare batterica attraverso interazioni tra le fibre della coda e il recettore (viola). Una volta sulla superficie cellulare, il fago viene irreversibilmente attaccato alla cellula batterica usando la piastra di base (nera) che viene spostata sulla parete cellulare dalla guaine contrattile (gialla). Il genoma dei fagi (rosso) entra quindi nella cellula e si integra nel genoma della cellula ospite.
Mentre la trasduzione batterica è un processo naturale, utilizzando la tecnologia moderna è stata manipolata per il trasferimento di geni nei batteri in laboratorio. Inserendo geni di interesse nel genoma di un fago lisogenico, come il fago, si è in grado di trasferire questi geni nei genomi dei batteri e di conseguenza esprimerli all'interno di queste cellule. Mentre altri metodi di trasferimento genico, come la trasformazione, utilizzano un plasmide per il trasferimento e l'espressione genica, l'inserimento del genoma del fago in quello del batterio ricevente non solo ha il potenziale per conferire nuovi tratti a questo batterio, ma consente anche mutazioni naturali e altri fattori dell'ambiente cellulare per alterare la funzione del gene trasferito.
Rispetto ad altri metodi di trasferimento genico orizzontale, come la coniugazione, la trasduzione è abbastanza flessibile nei criteri richiesti per le cellule donatrici e riceventi. Qualsiasi elemento genetico che può adattarsi all'interno del genoma del fago utilizzato può essere trasferito da qualsiasi ceppo di batteri donatori a qualsiasi ceppo di batteri riceventi purché entrambi siano permissivi al fago, richiedendo l'espressione dei necessari recettori fagici sulle superfici cellulari. Una volta che questo gene viene spostato fuori dal genoma del donatore e confezionato nel fago, può essere trasferito al ricevente. Dopo la trasduzione, è necessario selezionare per i batteri riceventi che contengono il gene di interesse devono essere selezionati per. Questo potrebbe essere fatto utilizzando un marcatore genetico, come un FLAG-tag o un polyhistidine-tag, per contrassegnare il gene di interesse, o la funzione intrinseca del gene, nel caso di geni che codificano per la resistenza agli antibiotici. Inoltre, la PCR potrebbe essere utilizzata per confermare ulteriormente il successo della trasduzione. Utilizzando primer per una regione all'interno del gene di interesse e confrontando il segnale con un controllo positivo, batteri che hanno il gene di interesse e un controllo negativo, batteri che hanno subito gli stessi passaggi della reazione di trasduzione senza fagi. Mentre la trasduzione batterica è uno strumento utile in biologia molecolare, ha e continua a svolgere un ruolo importante nell'evoluzione dei batteri, in particolare per quanto riguarda il recente aumento della resistenza agli antibiotici.
In questo esperimento, la trasduzione batterica è stata utilizzata per trasferire il gene che codifica per la resistenza all'ampicillina antibiotica dal ceppo W3110 di E.coli al ceppo J53 attraverso il batteriofago P1 (5). Questo esperimento consisteva in due fasi principali. In primo luogo, la preparazione del fago P1 contenente il gene di resistenza all'ampicillina dal ceppo donatore. In secondo luogo, il trasferimento di questo gene al ceppo ricevente per trasduzione con il fago P1 (Figura 1). Una volta effettuato, il successo del trasferimento del gene di resistenza all'ampicillina potrebbe essere determinato dalla qPCR (Figura 2). Se la trasduzione ha successo, il ceppo J53 di E. coli sarebbe resistente all'ampicillina e il gene che conferisce questa resistenza rilevabile dalla qPCR. In caso di insuccesta, non ci sarebbe alcun rilevamento del gene di resistenza all'ampicillina e l'ampicillina funzionerebbe ancora come un antibiotico efficace contro il ceppo J53.
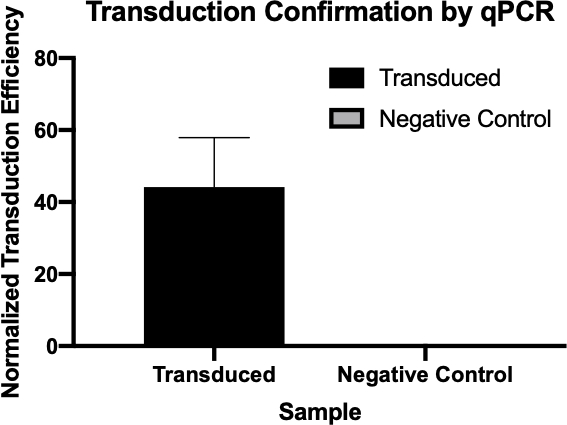
Figura 2: La conferma del successo della trasduzione da parte della qPCR. Confrontando i valori di Cq rilevati per il gene di interesse dalla reazione di trasduzione e dalla reazione di controllo negativa e normalizzando questi valori con un gene di pulizia, si è stati in grado di confermare che la trasduzione batterica ha avuto successo.
Procedure
1. Configurazione
- Prima di iniziare qualsiasi lavoro che coinvolga i microbi, sterilizzare lo spazio di lavoro utilizzando il 70% di etanolo. Utilizzare sempre i DPI necessari (camice da laboratorio e guanti).
- Assicurarsi che le piastre di agar LB con 1x ampicillina, soluzione di lisato fagico P1 disponibile in commercio, cloroformio, citrato di sodio 1 M, glicerolo e una scatola di punte di pipette di plastica pre-sterilizzate e diffusori di cellule siano a portata di mano.
- Preparare LB sterile in autoclave e utilizzarlo per produrre tre aliquote da 1 mL di soluzione salina LB.
- mM MgCl2 (952,11-2,3803 μg), 5 mM CaCl2 (11,098 mg), 0,1-0,2% di glucosio (100-200 μg)
- mM MgSO4 (12,0366 mg), 5 mM CaCl2 (11,098 mg)
- mM citrato di sodio (25,806 mg)
- Una volta terminato, sterilizzare tutte le superfici e i guanti con etanolo al 70% e lavarsi le mani.
2. Protocollo
- Preparazione del fago del donatore
- Preparare una coltura da 1 mL del ceppo E. coli del donatore W3110 in LB con 1x ampicillina coltivata durante la notte a 37 °C con aerazione e agitazione a 220 giri/min.
- Diluire questa coltura notturna 1:100 in 1 mL di LB fresco integrato con 10-25 mM MgCl2, 5 mM CaCl2e 0,1-0,2% di glucosio.
- Coltivare questa diluizione batterica a 37 °C per 2 ore con aerazione e agitazione a 220 giri/min.
- Una volta che queste cellule hanno raggiunto la fase di crescita logaritmica precoce (crescita evidente e leggera torbidità), aggiungere 40 μL di fago P1 disponibile in commercio e lasciare a 37 °C con aerazione e agitazione a 220 giri/min.
- Prima dell'aggiunta di fagi, la densità ottica misurata a 600 nm di queste cellule dovrebbe essere di circa 0,4 (6).
- Monitorare le cellule per 1-3 ore fino a quando la coltura non è stata lizzate.
- La lisi comporterà un aumento dei detriti cellulari e una notevole diminuzione della torbidità (cioè le cellule saranno considerate lizzate una volta che si è in grado di vedere attraverso la coltura).
- Aggiungere diverse gocce (50-100 μL) di cloroformio al lisato e mescolare per vortice.
- Il cloroformio sterilizza il lisito fagico uccidendo tutte le cellule donatrici rimanenti, lasciando solo fagi e aumentando il titolo di questo lisi.
- Centrifugare il lisi a 14.000 giri /min per 2 minuti per rimuovere i detriti e trasferire il surnatante in un tubo fresco.
- Aggiungere qualche goccia di cloroformio e conservare a 4 °C per non più di un giorno.
- Trasduzione
- Preparare una coltura da 1 mL del ceppo E. coli J53 ricevente coltivato durante la notte in LB a 37 °C con aerazione e agitazione a 220 giri/min.
- Trasferire 100 μL di fago di liscerato di fago donatore (2.1) in un tubo di microfuga da 1,5 ml e incubare con cappuccio aperto a 37 °C per 30 minuti.
- Questa incubazione consente a qualsiasi cloroformio rimanente nella soluzione di lisato P1 di evaporare prima di essere aggiunto alle cellule riceventi.
- Pellet delicatamente le celle di ceppo ricevente mediante centrifugazione a 6.000 giri /min per 5 minuti.
- Spese di sospensione di queste cellule in 300 μL di LB fresco contenente 100 mM MgSO4 e 5 mM CaCl2. (Il fago P1 richiede che il calcio sia infettivo).
- Impostare due reazioni utilizzando le cellule batteriche riceventi e il fago del donatore preparato lisato: 1) reazione di trasduzione che combina 100 μL ricevente J53 ceppo E. coli e 100 μL donatore fago lisato, e 2) controllo negativo che combina 100 μL ricevente J53 ceppo E. coli e 100 μL di LB contenente 100 mM MgSO4 e 5 mM CaCl2.
- Incubare a 37 °C per 30 minuti con agitazione a 220 giri/min.
- Aggiungere 1 mL LB e 200 μL 1M di citrato di sodio (pH=5,5) e incubare per 1 ora a 37 °C con agitazione a 220 giri/min.
- Il citrato viene utilizzato per ridurre l'infettività di P1 chelando con il calcio, prevenendo la lisi dei batteri riceventi.
- L'incubazione di questa soluzione consente l'espressione del marcatore di resistenza all'ampicillina.
- Celle a pellet per centrifugazione a 6.000 giri/min per 5 minuti.
- Pellet di celle spese in 100 μL di LB con 100 mM di citrato di sodio (pH 5,5). Vortice e piastra intera soluzione per entrambe le reazioni su due piastre lb agar.
- La piastra LB deve avere 1 Amp per il campione trasdotto e nessun Amp per il controllo negativo.
- La contaminazione dei fagi P1 su questa piastra richiede una ri-striatura prima che le scorte del congelatore possano essere preparate.
- Se il fago non viene rimosso, le colture coltivate da queste colonie non cresceranno se non in presenza di un chelatore di calcio.
- Prelevare circa 3-4 colonie da entrambe le piastre e strisciare di nuovo su due piastre di agar LB distribuite con 100 μL di 1 M di citrato di sodio (pH = 5,5).
- La piastra LB deve avere 1 Amp per il campione trasdotto e nessun Amp per il controllo negativo.
- Incubare le piastre a 37 °C durante la notte per consentire la crescita di colonie prive di fagi.
- Raccogliere colonie da entrambe le piastre e utilizzarle per coltivare colture notturne in 5 ml di LB a 37 °C con aerazione e agitazione a 220 giri/min.
- Isolare il DNA da queste colture mediante miniprep del DNA utilizzando 4,5 ml del volume totale della coltura.
- Eluire il DNA utilizzando 35 μL di acqua priva di nucleasi.
- Misurare la concentrazione risultante mediante Nanodrop. Il DNA puro genererà un rapporto di assorbanza (A260/280)di circa 1,8.
- Utilizzare i restanti 0,5 ml di ciascuna coltura per preparare 1 mL di scorte di glicerolo facendo una miscela 1:1 di glicerolo al 100% e coltura batterica.
- Conservare le scorte di glicerolo batterico a -80 °C.
3. Analisi dei dati e risultati
- Conferma della trasduzione mediante qPCR
- Preparare due miscele master qPCR per sei reazioni qPCR, tre utilizzando primer qPCR per il gene di resistenza all'ampicillina e le altre tre utilizzando primer qPCR per un gene di pulizia (14,5 μL per reazione): 12,5 μL di miscela tampone qPCR + 1 μL di primer in avanti + 1 μL di primer inverso.
- In questo esperimento, abbiamo usato SYBR Green master mix.
- I primer genetici di pulizia sono stati progettati per amplificare un segmento di DNA all'interno del gene batterico che codifica per la DNA girasi B (7).
- Per ogni reazione qPCR, combinare 100 μg di DNA da ciascuna reazione (10,5 μL) con 14,5 μL di miscela master qPCR.
- Utilizzando una macchina qPCR e il protocollo di termociclizzazione elencato nella Tabella 1, è stata misurata l'amplificazione per la resistenza all'ampicillina e i geni di pulizia per tutte e sei le reazioni.
- I valori di Cq generati da qPCR sono stati utilizzati per calcolare l'efficienza di trasduzione normalizzata del gene di resistenza all'ampicillina (Figura 3), confermando il successo della trasduzione del gene di resistenza all'ampicillina.
- Il valore Cq, o valore di quantificazione del ciclo, di un campione è il primo numero di ciclo PCR al quale viene rilevato un segnale che supera la soglia di fondo. I valori Cq bassi corrispondono a una sequenza di destinazione maggiore, viceversa.
- L'efficienza di trasduzione normalizzata di un gene all'interno di un campione può essere calcolata utilizzando questi valori di Cq sottraendo il valore del gene di pulizia da quello del gene bersaglio, generando un valore ΔCq, che può essere utilizzato per calcolare l'efficienza di trasduzione normalizzata di 2(-ΔCq).
- Preparare due miscele master qPCR per sei reazioni qPCR, tre utilizzando primer qPCR per il gene di resistenza all'ampicillina e le altre tre utilizzando primer qPCR per un gene di pulizia (14,5 μL per reazione): 12,5 μL di miscela tampone qPCR + 1 μL di primer in avanti + 1 μL di primer inverso.
| Temperatura | Ore | |
| Denaturazione | 94 °C | 2 minuti |
| 40 cicli: | ||
| Denaturazione | 94 °C | 15 secondi |
| Ricottura, estensione e fluorescenza letta | 60 °C o 5 °C al di sotto del primer Tm più basso | 1 min |
Tabella 1: Protocollo di termociclica qPCR
I batteri possono adattarsi rapidamente a un ambiente in rapida evoluzione scambiando materiale genetico e un modo in cui possono farlo è attraverso la trasduzione, lo scambio di materiale genetico mediato da virus batterici. Un batteriofago, spesso abbreviato in fago, è un tipo di virus che infetta i batteri attaccandosi prima alla superficie dell'ospite e poi iniettando il suo DNA nella cellula batterica. Quindi degrada il DNA della cellula ospite e replica il suo genoma virale, mentre dirotta il macchinario della cellula per sintetizzare molte copie delle sue proteine. Queste proteine fagiche si auto-assemblano e impacchettare i genomi dei fagi per formare più progenie. Tuttavia, a causa della bassa fedeltà del meccanismo di confezionamento del DNA, occasionalmente, il fago impacchettare frammenti di DNA batterico nel capside del fago. Dopo aver indotto la lisi dell'ospite, la progenie del fago viene rilasciata e, una volta che tale fago infetta un'altra cellula ospite, trasferisce il frammento di DNA del suo ospite precedente. Questo può quindi ricombinarsi e diventare permanentemente incorporato nel cromosoma del nuovo ospite, mediando così il trasferimento genico tra i due batteri.
Per effettuare la trasduzione dei fagi in laboratorio è necessario un ceppo donatore che contiene un gene di interesse, un ceppo ricevente che ne è privo, un fago che può infettare entrambi i ceppi e un metodo per selezionare i batteri trasdotti. Nella maggior parte dei casi, questo sarà un mezzo di crescita solido selettivo che supporta la crescita di batteri trasdotti ma inibisce la crescita di quelli non trasdotti. Per iniziare, il ceppo donatore che contiene il gene di interesse viene coltivato in un terreno di crescita liquido. Quando tutti i batteri si dividono attivamente nella fase logaritmica della loro crescita, la coltura viene inoculata con il fago bersaglio. Dopo tre o quattro ore di incubazione, quando quasi tutti i batteri hanno lizzata e rilasciato le particelle fagiche, il lisito fagico donatore viene inoculato in una coltura appena coltivata del ceppo batterico ricevente. Dopo una breve incubazione di un'ora, la coltura dovrebbe ora contenere una miscela di cellule batteriche trasdotte e non trasdotte e questa viene sottoposta a screening per le cellule trasdotte diffondendo una frazione della sospensione su un appropriato mezzo di crescita solido selettivo. Dopo un'ulteriore incubazione, le cellule trasdotte dovrebbero crescere e moltiplicarsi per produrre colonie visibili. Queste colonie possono quindi essere selezionate per ulteriori analisi utilizzando una varietà di metodi per confermare ulteriormente la trasduzione di successo, come la PCR della colonia, il sequenziamento del DNA o la PCR quantitativa.
Prima di iniziare la procedura, indossare qualsiasi dispositivo di protezione individuale appropriato, tra cui un camice da laboratorio e guanti. Quindi, sterilizzare lo spazio di lavoro con etanolo al 70% e pulire la superficie.
Successivamente, preparare tre aliquote da un millilitro di soluzione salina LB. Ora, preparare una coltura di ceppo donatore aggiungendo 100 microlitri di E. coli a una fiala conica da 15 millilitri contenente cinque millilitri di terreno di coltura LB con 500 microgrammi di ampicillina. Quindi, coltiva la coltura durante la notte a 37 gradi Celsius con aerazione e agitazione a 220 giri / min. Il giorno dopo, pulire il piano di lavoro con il 70% di etanolo prima di rimuovere la coltura dall'incubatore vibrante. Quindi, diluire la coltura notturna a 100 aggiungendo 10 microlitri di ceppo donatore a 990 microlitri di LB fresco integrato con soluzione salina.
Lasciare che la diluizione batterica cresca a 37 gradi Celsius per due ore con aerazione e agitazione a 220 giri / min. Una volta che le cellule hanno raggiunto la fase iniziale del log, rimuovere la coltura dall'incubatore, aggiungere 40 microlitri di fago P1 alla coltura e incubare di nuovo. Continuare a monitorare le cellule per una o tre ore fino a quando la coltura non si è lssata. Quindi, aggiungere da 50 a 100 microlitri di cloroformio al lisi e mescolare vorticosamente. Quindi, centrifugare il l'lisi per rimuovere i detriti e trasferire il surnatante in un tubo fresco. Aggiungere alcune gocce di cloroformio al surnatante e conservarlo a quattro gradi Celsius per non più di un giorno.
Per iniziare la procedura di trasduzione, ottenere una coltura di un millilitro del ceppo ricevente. Quindi, trasferire 100 microlitri di fago disalato donatore in un tubo microcentrifuga da 1,5 millilitri e incubarlo a 37 gradi Celsius con il cappuccio aperto per 30 minuti per consentire all'eventuale cloroformio rimanente di evaporare. Mentre il fago del donatore il lidato incuba, pellet le cellule del ceppo ricevente tramite centrifugazione delicata. Scartare il surnatante e risusegnare il pellet cellulare in 300 microlitri di LB fresco contenenti 100 solfato di magnesio millimolare e cloruro di calcio di cinque millimolari.
Successivamente, impostare la reazione di trasduzione combinando 100 microlitri del ceppo ricevente e 100 microlitri del fago del donatore lizzato in un tubo microcentrifuga. Quindi, impostare il controllo negativo combinando 100 microlitri del ceppo ricevente e 100 microlitri del LB con solfato di magnesio e cloruro di calcio. Dopo l'incubazione, aggiungere 200 microlitri di un citrato di sodio molare e un millilitro di LB a entrambi i tubi e mescolare delicatamente il pipettaggio su e giù. Quindi, dopo che i tubi sono stati incubati per un'ora, pelletttare delicatamente le cellule tramite centrifugazione.
Dopo la centrifugazione, scartare il surnatante e risuscidere le cellule pellettizzate in 100 microlitri di LB con citrato di sodio 100 millimolare. Vortice delle soluzioni e pipettare l'intero campione trasdotto su una piastra di agar LB con ampicillina 1X. Infine, pipettare l'intero volume della miscela di cellule di controllo negativo su una piastra di agar LB senza ampicillina. Dopo aver incubato le piastre durante la notte a 37 gradi Celsius, utilizzare una punta di pipetta sterile per raccogliere da tre a quattro colonie dalla piastra di trasduzione e strisciarle su una nuova piastra di agar LB contenente 1X ampicillina e 100 microlitri di un citrato di sodio molare. Ripetere questo metodo di placcatura per il controllo negativo su un'altra piastra di agar LB contenente solo 100 microlitri di un citrato di sodio molare. Quindi, incubare le piastre a 37 gradi Celsius durante la notte per consentire alle colonie prive di fagi di crescere.
Il giorno successivo, pulire il piano di lavoro con il 70% di etanolo prima di rimuovere le piastre dall'incubatrice. Utilizzando una punta di pipetta sterile, prelevare tre colonie dalla piastra di trasduzione e aggiungerle ciascuna a un tubo separato contenente cinque millilitri di supporti LB. Quindi, selezionare tre colonie dalla piastra di controllo negativa e aggiungerle a un altro tubo contenente cinque millilitri di supporti LB. Coltiva le colture durante la notte a 37 gradi Celsius con aerazione e agitazione a 220 giri / min. Dopo aver sterilizzato il piano di lavoro come precedentemente dimostrato, utilizzare un kit miniprep DNA per isolare il DNA da 4,5 millilitri di ciascuna coltura secondo le istruzioni del produttore. Quindi, eluire il DNA con 35 microlitri di acqua priva di nucleasi e misurare la concentrazione risultante mediante spettrofotometro da laboratorio. Infine, preparare le scorte di glicerolo aggiungendo i restanti 0,5 millilitri di entrambe le soluzioni batteriche a 0,5 millilitri di glicerolo al 100%.
Per confermare la trasduzione, preparare prima due miscele master qPCR per 24 reazioni qPCR. Per il primo master mix, aggiungere 150 microlitri di miscela tampone qPCR a un tubo microcentrifuga e 12 microlitri ciascuno di un primer avanti e indietro progettato per amplificare il gene della resistenza all'ampicillina. Quindi, preparare un secondo mix master qPCR aggiungendo 150 microlitri di mix master qPCR a un tubo microcentrifuga e quindi aggiungendo 12 microlitri ciascuno di un primer avanti e un primer inverso progettati per amplificare un gene di pulizia.
Per ogni reazione qPCR, combinare 100 microgrammi di DNA sperimentale da ogni reazione con 14,5 microlitri di miscela master qPCR. Ora, prepara le reazioni rimanenti come precedentemente dimostrato. Trasferire le reazioni a un termociclatore preriscaldato a 94 gradi Celsius e quindi avviare il programma. Infine, utilizzare i valori di quantificazione del ciclo, o Cq, generati da qPCR per calcolare l'efficienza di trasduzione normalizzata del gene di resistenza all'ampicillina.
I valori di quantificazione del ciclo, o Cq, per i geni di interesse sono stati tabulati per ciascuno dei controlli negativi e dei campioni trasdotti. Valori Cq bassi, in genere inferiori a 29 cicli, come i campioni trasdotti in questo esempio indicano quantità elevate della sequenza target.
Un gene di pulizia, anche qui tabulato, viene utilizzato come controllo del carico per normalizzare la quantità di DNA in ogni reazione e come controllo positivo per garantire che la qPCR funzioni. A condizione che vengano caricate le stesse quantità del gene di pulizia, si trova relativamente alla stessa velocità in ciascun campione.
Successivamente, per calcolare il valore delta Cq per ciascun campione, sottrarre il valore Cq del gene housekeeping per ciascun campione dal valore Cq del gene bersaglio corrispondente. Ad esempio, il delta Cq del primo controllo negativo è 13,54. Quindi, utilizzare questo valore per calcolare l'efficienza di trasduzione normalizzata di ciascun campione utilizzando la formula mostrata qui. Infine, è possibile calcolare l'efficienza media di trasduzione normalizzata per ciascun gruppo di campioni.
Subscription Required. Please recommend JoVE to your librarian.
Applications and Summary
Il trasferimento di geni da e verso i batteri da parte del batteriofago, pur essendo un processo naturale, si è rivelato estremamente utile per una moltitudine di scopi di ricerca. Mentre sono possibili altri metodi di trasferimento genico come la trasformazione e la coniugazione, la trasduzione utilizza in modo univoco i batteriofagi; non solo consentendo l'integrazione genica nel genoma dell'ospite, ma anche la consegna di geni a più batteri che non sono suscettibili ad altri metodi. Questo processo, sebbene particolarmente utile in laboratorio, è stato utilizzato anche nel campo recentemente emergente della terapia genica, più specificamente nella terapia genica alternativa, una strategia terapeutica che utilizza i batteri per fornire terapie ai tessuti bersaglio, molti dei quali non sono suscettibili ad altri metodi di consegna e hanno molta rilevanza clinica (8,9).
Subscription Required. Please recommend JoVE to your librarian.
References
- Lederberg J, Lederberg E.M., Zinder, N.D., et al. Recombination analysis of bacterial heredity. Cold Spring Harbor symposia Quantitative Biol. 1951;16:413-43.
- Duckworth DH. "Who Discovered Bacteriophage?". Bacteriology Reviews. 1976;40:793-802.
- Yap ML, Rossman, M.G. Structure and Function of Bacteriophage T4. Future Microbiol. 2014;9:1319-27.
- Sulakvelidze A, Alavidze, Z., Morris, J. G. Bacteriophage Therapy Antimicrobial Agents and Chemotherapy 2001;45(3):649-59.
- Moore S. Sauer:P1vir phage transduction 2010 [Available from: https://openwetware.org/wiki/Sauer:P1vir_phage_transduction].
- Kobayashi A, et al. Growth Phase-Dependent Expression of Drug Exporters in
- Escherichia coli and Its Contribution to Drug Tolerance. Journal of Bacteriology. 2006;188(16):5693-703.
- Rocha D, Santos, CS, Pacheco LG. Bacterial reference genes for gene expression studies by RT-qPCR: survey and analysis. Antonie Van Leeuwenhoek. 2015;108:685-93.
- Pálffy R. et al. Bacteria in gene therapy: bactofection versus alternative gene therapy. Gene Ther. 2006 13:101-5.
- O'Neill JM, et al. Intestinal delivery of non-viral gene therapeutics: physiological barriers and preclinical models. Drug Discovery Today. 2011;16:203-2018.