



Overview
출처: 알렉산더 S. 골드1, 토냐 M. 콜핏스1
1 미생물학학과, 보스턴 대학교 의과대학, 국립 신흥 감염 질병 연구소, 보스턴, MA
트랜스듀션은 박테리아 간의 유전적 교류의 한 형태로, 배니오파지 또는 파지, 독점적으로 대핵 생물을 감염시키는 바이러스의 종류입니다. 이 형태의 DNA 전달은 파지를 통해 박테리아에서 다른 박테리아로, 1951년에 노턴 진더와 조슈아 레더그(Joshua Ledererg)에 의해 발견되었으며, 이들은 이 과정을 "트랜스듀션"(1)이라고 지칭했다. 박테리오파지는 1915년 영국 세균학자 프레드릭 투르트(Frederick Twort)에 의해 처음 발견되었으며, 1917년 프랑스계 캐나다의 미생물학자 펠릭스 데렐(Felix d'Herelle,2)에 의해 독립적으로 발견되었다. 그 이후로, 이러한 파지의 구조와 기능은 널리 특징지어지고 있다 (3), 두 개의 클래스로 이러한 파지를 분할. 이 클래스의 첫 번째는 감염시 숙주 박테리아 내에서 증식하는 lytic 파지, 세균 대사를 방해, 세포를 lysing, 그리고 자손 파지를 해제 (4). 이 항균 활성과 항생제 내성 박테리아의 보급증가의 결과로, 이러한 lytic 파지는 최근 항생제에 대한 대체 치료로 유용함을 입증했다. 이들 클래스의 두 번째는 리액 사이클을 통해 숙주 내에서 증식하거나 게놈이 숙주(그림 1)에 통합되는 정지 상태를 입력할 수 있는 리소겐성 파지이며, 파지 생산능력이 다세대(4)에 유도될 수 있다.
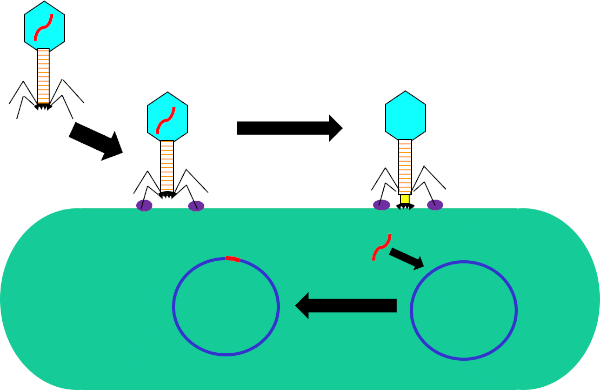
그림 1: 박테리오파지에 의한 숙주 세포의 감염. 꼬리 섬유와 수용체 (보라색) 사이의 상호 작용을 통해 세균 세포 벽에 파지에 의한 흡착. 일단 세포 표면에, 파지는 수축 칼집 (노란색)에 의해 세포벽으로 이동하는 베이스 플레이트 (black)를 사용하여 박테리아 세포에 돌이킬 수 없이 부착된다. 파지 게놈(red)은 세포에 들어가 숙주 세포 게놈에 통합한다.
세균성 트랜스포팅은 자연적으로 발생하는 과정이지만, 현대 기술을 사용하여 실험실 환경에서 유전자를 박테리아로 옮기기 위해 조작되었습니다. 파지와 같은 리존파지의 게놈에 관심 있는 유전자를 삽입함으로써, 이러한 유전자를 박테리아의 게놈으로 옮기고 결과적으로 이러한 세포 내에서 이를 표현할 수 있다. 변형과 같은 유전자 전달의 다른 방법은, 유전자 전달 및 발현을 위해 플라스미드를 사용하지만, 파지 게놈을 받는 박테리아의 것으로 삽입하는 것은 이 박테리아에 새로운 특성을 부여할 뿐만 아니라, 또한 전송된 유전자의 기능을 바꾸기 위하여 세포 환경의 자연적으로 발생하는 돌연변이 및 그밖 요인을 허용합니다.
유도와 같은 수평 유전자 전달의 다른 방법에 비해, 전달은 기증자 및 수령인 세포에 필요한 기준에 상당히 유연하다. 사용되는 파지의 게놈 내부에 들어갈 수 있는 모든 유전 적 원소는 기증자 박테리아의 변형에서 모두 파지에 허용되는 한 받는 사람 박테리아의 변형으로 옮겨질 수 있으며 세포 표면에 필요한 파지 수용체의 발현이 필요합니다. 일단 이 유전자는 기증자 게놈에서 이동하고 파지로 포장되면, 수신자에게 전달될 수 있습니다. 트랜스듀션에 따라 관심 유전자를 포함하는 수신자 박테리아를 선택해야 한다. 이것은 항생 저항을 위해 인코딩하는 유전자의 경우에, 관심의 유전자, 또는 유전자의 본질적인 기능을 표시하기 위하여, FLAG-tag 또는 polyhistidine-tag와 같은 유전 마커의 사용에 의해 행해질 수 있었습니다. 또한 PCR을 사용하여 성공적인 트랜스듀션을 확인할 수 있습니다. 관심 있는 유전자 내의 영역에 프라이머를 사용하고 신호를 긍정적 인 대조군, 관심 유전자가 있는 박테리아 및 음의 대조군, 파지 없이 트랜스유도 반응과 동일한 단계를 겪은 박테리아를 사용한다. 세균성 트랜스포메이션은 분자 생물학에 유용한 도구이지만, 특히 최근 항생제 내성의 부상과 관련하여 박테리아의 진화에 중요한 역할을 하고 있습니다.
본 실험에서, 세균성 트랜스포칭은 P1 박테리오파지(5)를 통해 E.coli의 W3110 균주로부터 항생제 암피실린에 대한 내성을 위한 유전자 인코딩 유전자를 전송하는 데 사용되었다. 이 실험은 두 가지 주요 단계로 구성되었습니다. 첫째, 기증자 균주로부터 암피실린 내성 유전자를 함유하는 P1 파지의 제제. 둘째, P1 파지와의 트랜스듀션에 의한 수신자 균주에 대한 이 유전자의 전달(도 1). 일단 수행되면, 암피실린 저항 유전자의 성공적인 전송은 qPCR에 의해 결정될 수 있었다(도 2). 트랜스포팅이 성공하면 대장균의 J53 균주는 암피실린에 내성이 있으며, qPCR에 의해 검출가능한 이 저항을 부여하는 유전자가 될 것이다. 실패하는 경우에, 암피실린 저항 유전자의 아무 검출도 없을 것이고 ampicillin는 J53 긴장에 대하여 효과적인 항생제로 아직도 작동할 것입니다.
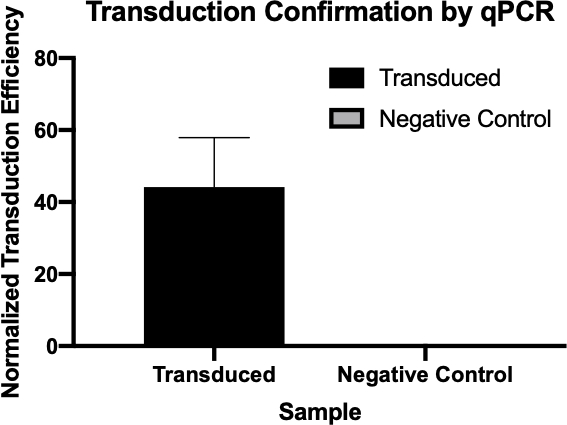
그림 2: qPCR에 의한 성공적인 트랜스듀션확인. 트랜스듀션 반응 및 음성 대조반응으로부터 관심 유전자에 대해 검출된Cq 값을 비교하고, 가사 유전자에 대하여 이러한 값을 정상화함으로써, 세균성 전이 성공했다는 것을 확인할 수 있었다.
Procedure
1. 셋업
- 미생물과 관련된 작업을 시작하기 전에 70 %의 에탄올을 사용하여 작업 공간을 살균하십시오. 항상 필요한 PPE (실험실 코트와 장갑)를 사용합니다.
- 1x 암피실린, 시판 가능한 P1 파지 용액, 클로로폼, 1M 나트륨 구산염, 글리세롤, 미리 멸균된 플라스틱 파이펫 팁 및 셀 스프레더 상자가 있는 LB agar 플레이트가 가까이 에 있는지 확인하십시오.
- 오토클레이브를 사용하여 멸균 LB를 준비하고 이를 사용하여 LB 소금 용액 의 3mL 알리쿼트 3개 만들기에 사용하십시오.
- mM MgCl2 (952.11-2.3803 μg), 5 mM CaCl2 (11.098 mg), 0.1-0.2 % 포도당 (100-200 μg)
- mM MgSO4 (12.0366 mg), 5 mM CaCl2 (11.098 mg)
- mM 나트륨 구연산나트륨 (25.806 mg)
- 완성되면 모든 표면과 장갑을 70% 에탄올로 살균하고 손을 씻으실 수 있습니다.
2. 프로토콜
- 기증자 파지 리세테 준비
- 1mL 의 기증자 W3110 균주 대장균을 LB에서 1x 암피실린으로 37°C에서 하룻밤 동안 재배하고 220 rpm에서 흔들어 줍니다.
- 이 하룻밤 문화를 희석 1:100 신선한 LB의 1 mL에 10-25 mM MgCl2,5 mM CaCl2,그리고 0.1-0.2% 포도당.
- 이 세균 희석을 37°C에서 2시간 동안 증가시키고 220rpm에서 흔들어 보입니다.
- 이러한 세포가 초기 로그산학 성장 단계(눈에 띄는 성장과 약간의 탁도)에 도달하면, 시판되는 P1 파지의 40μL을 추가하고 37°C에서 폭기와 220 rpm에서 흔들어 둡니다.
- 파지 첨가 전에, 이들 세포의 600 nm에서 측정된 광학 밀도는 약 0.4(6)여야 한다.
- 배양이 용해 될 때까지 1-3 시간 동안 셀을 모니터링합니다.
- Lysis는 세포 이물질이 증가하고 탁도가 현저한 감소(즉, 세포가 배양을 통해 볼 수 있게 되면 lysed 것으로 간주됩니다).
- 용액에 클로로폼의 여러 방울 (50-100 μL)을 추가하고 소용돌이에 의해 혼합합니다.
- 클로로폼은 남은 기증자 세포를 죽이고 파지만 남기고 이 리세이트의 티터를 증가시킴으로써 파지 리세이스를 살균합니다.
- 원심분리기는 14,000rpm에 2분 동안 이물질을 제거하고 상퍼를 신선한 튜브로 이송합니다.
- 클로로폼 몇 방울을 넣고 4°C에 보관하여 하루 이상 보관하십시오.
- 변환
- 37°C에서 LB에서 하룻밤 재배한 수취인 J53 균주 대장균의 1mL 배양을 준비하여 220 rpm에서 흔들어 줍니다.
- 기증자 파지 용해(2.1)의 100 μL을 1.5mL 마이크로퍼지 튜브로 옮기고 30분 동안 37°C에서 캡을 열어 배양합니다.
- 이 배양은 수신자 세포에 추가되기 전에 증발하는 P1 lysate 용액에 있는 모든 나머지 클로로폼을 허용합니다.
- 부드럽게 펠릿 받는 사람 변형 세포에 의해 원심 분리에 의해 6,000 rpm 에 5 분.
- 이러한 세포를 100m MgSO4 및 5mM CaCl2를함유한 신선한 LB300 μL에서 재보중단한다. (P1 파지는 전염성이 있어야 합니다).
- 수신자 박테리아 세포를 사용하여 2개의 반응을 설정하고 준비된 기증자 파지 용액: 1) 100 μL 수신자 J53 균주 E. 대장균과 100 μL 기증자 파지 용해, 및 2) 100 μL 수신자 J53 균주 E. 대장균 및100 μL의 LB를 포함하는 100 μL 감산 염기 및 100 μL를 결합한 음성 대조군
- 37°C에서 30분간 인큐베이션을 하고 220rpm에서 흔들리면 됩니다.
- 1mL LB 와 200 μL 1M 나트륨 구연산나트륨(pH=5.5)을 넣고 37°C에서 1시간 동안 배양하고 220rpm에서 흔들어 앉습니다.
- 체연산은 칼슘으로 다수화하여 P1의 감염성을 감소시켜 받는 박테리아의 용해를 방지하는 데 사용됩니다.
- 이 용액의 배큐어는 암피실린 저항 마커의 발현을 허용한다.
- 펠릿 세포는 5 분 동안 6,000 rpm에서 원심 분리에 의해 세포.
- 100mM 나트륨 구연산염(pH 5.5)으로 LB의 100 μL에서 세포 펠릿을 재중단한다. 두 LB 천 플레이트에 대한 두 반응에 대한 소용돌이 및 플레이트 전체 용액.
- LB 플레이트는 변환된 샘플에 대해 1x Amp가 있어야 하며 음수 제어를 위한 앰프가 없어야 합니다.
- 이 플레이트의 P1 파지 오염은 냉동고 재고를 준비하기 전에 다시 줄무늬가 필요합니다.
- 파지를 제거하지 않으면, 칼슘 첼라토르가 없는 한, 이러한 식민지에서 자란 문화는 성장하지 않을 것입니다.
- 플레이트에서 약 3-4개의 콜로니를 선택하고 구연산나트륨 1M의 100 μL(pH=5.5)로 확산되는 두 개의 LB 한천 판에 다시 줄무늬를 선택합니다.
- LB 플레이트는 변환된 샘플에 대해 1x Amp가 있어야 하며 음수 제어를 위한 앰프가 없어야 합니다.
- 파지가 없는 식민지가 성장할 수 있도록 하룻밤 사이에 37°C의 플레이트를 배양합니다.
- 두 접시에서 식민지를 선택하고 폭기와 함께 37 °C에서 LB의 5 mL에서 하룻밤 문화를 성장하고 220 rpm에서 흔들어 그들을 사용합니다.
- 총 배양량의 4.5mL를 사용하여 DNA 미니프렙에 의해 이러한 배양으로부터 DNA를 분리한다.
- 뉴클레아제없는 물의 35 μL을 사용하여 엘루트 DNA.
- 나노 드롭에 의해 결과 농도를 측정합니다. 순수 DNA는 약 1.8의 흡광도 비(A260/280)를생성합니다.
- 100% 글리세롤과 세균 배양의 1:1 혼합물을 만들어 1mL 글리세롤 주식을 제조하기 위해 각 배양의 나머지 0.5 mL을 사용한다.
- 세균글리세롤 재고를 -80°C에 저장합니다.
3. 데이터 분석 및 결과
- qPCR에 의한 트랜스듀스 확인
- 6qPCR 반응에 대한 2개의 qPCR 마스터 믹스, 암피실린 저항 유전자에 대한 qPCR 프라이머를 사용하는 3개, 다른 3개들은 가사 유전자에 대한 qPCR 프라이머(반응당 14.5 μL): 12.5 μL qPCR 버퍼 믹스 + 1 μL 포워드 프라이머 + 1μL 포워드 프라이머 + 1μL 포워드 프라이머 + 1μL 포워드 프라이머 + 1μL 포워드 프라이머 + 1μL 포워드 프라이머 + 1μL 포워드 프라이머 + 1μL 포워드 프라이머 + 1μL 포워드 프라이머 + 1μL 포워드 프라이머 + 1μL 포워드 프라이머 + 1μL 포워드 프라이머 + 1μL 포워드 프라이머 + 1μL 포워드 프라이머 + 1μL 포워드 프라이머 + 1μL 포워드 프라이머 + 1μL 포워드 프라이머 + 1μL 포워드 프라이머 + 1μL 포워드 프라이머 + 1μL 포워드 프라이머 + 1μL 포워드 프라이머 + 1μL 포워드 프라이머 + 1μL 포워드 프라이머 + 1μL 포워드 프라이머 + 1μL 포워드 프라이머 + 1μL 포워드 프라이머 + 1
- 이 실험에서, 우리는 SYBR 그린 마스터 믹스를 사용했다.
- 가사 유전자 프라이머는 DNA 자이라제 B (7)에 대한 인코딩 세균 유전자 내의 DNA 세그먼트를 증폭하도록 설계되었습니다.
- 각 qPCR 반응에 대해 각 반응(10.5 μL)에서 100 μg의 DNA를 qPCR 마스터 믹스의 14.5 μL로 결합합니다.
- 표 1에 나열된 qPCR 기계 및 써모사이클링 프로토콜을 사용하여, 증폭은 6개의 반응 모두에 대한 암피실린 저항성 및 하우스키핑 유전자를 위해 측정되었다.
- qPCR에 의해 생성된 Cq 값은 암피실린 저항 유전자(도 3)의 정규화된 트랜스듀션 효율을 계산하기 위해 사용되었고, 암피실린 저항 유전자의 성공적인 트랜스듀션을 확인시켰다.
- 샘플의 Cq 값 또는 사이클 정량화 값은 신호가 백그라운드 임계값을 초과한 초기 PCR 사이클 번호입니다. 낮은 Cq 값은 더 많은 대상 시퀀스에 해당하며, 그 반대의 경우도 마찬가지입니다.
- 시료 내 유전자의 정규화된 트랜스듀션 효율은 표적 유전자로부터 가사유전자의 값을 빼서 이러한 Cq 값을 사용하여 계산할 수 있으며, ΔCq 값을 생성하여 정규화된 트랜스듀션 효율을2(-ΔCq)로계산하는 데 사용될 수 있다.
- 6qPCR 반응에 대한 2개의 qPCR 마스터 믹스, 암피실린 저항 유전자에 대한 qPCR 프라이머를 사용하는 3개, 다른 3개들은 가사 유전자에 대한 qPCR 프라이머(반응당 14.5 μL): 12.5 μL qPCR 버퍼 믹스 + 1 μL 포워드 프라이머 + 1μL 포워드 프라이머 + 1μL 포워드 프라이머 + 1μL 포워드 프라이머 + 1μL 포워드 프라이머 + 1μL 포워드 프라이머 + 1μL 포워드 프라이머 + 1μL 포워드 프라이머 + 1μL 포워드 프라이머 + 1μL 포워드 프라이머 + 1μL 포워드 프라이머 + 1μL 포워드 프라이머 + 1μL 포워드 프라이머 + 1μL 포워드 프라이머 + 1μL 포워드 프라이머 + 1μL 포워드 프라이머 + 1μL 포워드 프라이머 + 1μL 포워드 프라이머 + 1μL 포워드 프라이머 + 1μL 포워드 프라이머 + 1μL 포워드 프라이머 + 1μL 포워드 프라이머 + 1μL 포워드 프라이머 + 1μL 포워드 프라이머 + 1μL 포워드 프라이머 + 1μL 포워드 프라이머 + 1
| 온도 | 시간 | |
| 변성 | 94 °C | 2분 |
| 40 사이클: | ||
| 변성 | 94 °C | 15초 |
| 아닐링, 확장 및 형광 읽기 | 최저 프라이머Tm 아래 60°C 또는 5°C | 1분 |
표 1: qPCR 써모사이클링 프로토콜
박테리아는 유전 물질을 교환하여 빠르게 변화하는 환경에 빠르게 적응할 수 있고 이것을 할 수 있는 한 가지 방법은 세균바이러스에 의해 매개되는 유전 물질의 교환인 트랜스듀션을 통해서입니다. 종종 파지에 축약 된 박테리아는 먼저 숙주의 표면에 부착 한 다음 박테리아 세포에 DNA를 주입하여 박테리아를 감염시키는 바이러스의 유형입니다. 그런 다음 숙주 세포의 DNA를 분해하고 바이러스 게놈을 복제하는 동시에 세포의 기계를 납치하여 단백질의 많은 사본을 합성합니다. 이 파지 단백질은 그 때 자기 조립하고 다중 자손형성을 형성하기 위하여 파지 게놈을 포장합니다. 그러나 DNA 포장 메커니즘의 낮은 충실도로 인해 파지는 박테리아 DNA의 단편을 파지 캡시드로 포장합니다. 숙주의 리시스를 유도한 후 파지 자손이 방출되고, 일단 파지가 다른 숙주 세포를 감염하면 이전 숙주의 DNA 단편을 전달합니다. 이것은 그 때 다시 결합하고 새로운 호스트의 염색체로 영구적으로 통합될 수 있고, 따라서 2개의 박테리아 사이 유전자 전송을 중재할 수 있습니다.
실험실에서 파지 변환을 수행하기 위해서는 관심 유전자를 포함하는 기증자 균주, 부족한 받는 사람 균주, 균주를 모두 감염시킬 수 있는 파지 및 변형된 박테리아를 선택하는 방법이 필요합니다. 대부분의 경우, 이것은 트랜스듀스 된 박테리아의 성장을 지원하지만 비 유도 된 박테리아의 성장을 억제하는 선택적 고체 성장 미디어가 될 것입니다. 시작하려면, 관심 유전자를 포함하는 기증자 균주는 액체 성장 배지에서 배양된다. 모든 박테리아가 성장의 로그 단계에서 활발하게 분할될 때, 배양은 표적 파지로 접종된다. 3~4시간 동안 인큐베이션을 한 후, 거의 모든 박테리아가 파지 입자를 분해하고 방출했을 때, 기증자 파지 리스는 받는 사람의 세균균균의 갓 재배된 배양으로 접종됩니다. 1시간의 간략한 잠복 후, 배양은 이제 변형및 비유도 세균 세포의 혼합물을 포함해야 하며, 이것은 적절한 선택적 고체 성장 매체에 서스펜션의 일부를 확산시킴으로써 트랜스포제드된 세포를 위해 선별된다. 추가 인큐베이션시, 유도된 세포는 눈에 보이는 식민지를 산출하기 위하여 성장하고 곱해야 합니다. 이러한 식민지는 식민지 PCR, DNA 염기서열 분석 또는 정량PCR과 같은 성공적인 전염을 더욱 확인하기 위해 다양한 방법을 사용하여 추가 분석을 위해 선택될 수 있다.
절차를 시작하기 전에 실험실 코트와 장갑을 포함한 적절한 개인 보호 장비를 착용하십시오. 다음으로, 70% 에탄올로 작업 공간을 살균하고 표면을 닦아냅니다.
그 후, LB 염액의 3개의 1밀리리터 알리쿼트를 준비하십시오. 지금, 암피실린의 500 마이크로그램을 가진 LB 성장 매체의 5밀리리터를 포함하는 15 밀리리터 원내 유리병에 대장균의 100 마이크로리터를 추가하여 기증자 긴장 문화를 준비합니다. 그런 다음, 220 rpm에서 폭기와 흔들림과 함께 섭씨 37도에서 하룻밤 문화를 성장. 다음 날, 흔들리는 인큐베이터에서 문화를 제거하기 전에 70 %의 에탄올로 벤치 상단을 닦아. 다음으로, 10 마이크로리터의 기증자 균주를 소금 용액으로 보충한 990 마이크로리터에 첨가하여 하룻밤 문화를 1~100으로 희석시켰다.
세균 희석이 2시간 동안 섭씨 37도에서 320rpm에서 흔들도록 허용합니다. 세포가 초기 로그 단계에 도달하면 인큐베이터에서 배양을 제거하고 배양에 P1 파지 의 40 마이크로 리터를 추가하고 다시 배양하십시오. 배양이 용해 될 때까지 1 ~ 3 시간 동안 세포를 계속 모니터링하십시오. 다음으로, 50~100마이크로리터의 클로로폼을 리세이트에 넣고 소용돌이에 섞습니다. 그런 다음, 리세이트 원심분리하여 파편을 제거하고 상체를 신선한 튜브로 옮기습니다. 상류체에 클로로폼 몇 방울을 넣고 하루 이상 섭씨 4도에 보관하십시오.
트랜스듀션 절차를 시작하려면 받는 사람 변형의 1 밀리리터 배양을 얻습니다. 다음으로, 기증자 파지 용액 100 마이크로리터를 1.5 밀리리터 미세센심분리기 튜브로 옮기고 30분 동안 캡을 열어 37도에서 배양하여 남은 클로로폼이 증발할 수 있도록 한다. 기증자 파지가 인큐베이션하는 동안, 펠릿은 부드러운 원심 분리를 통해 받는 사람 변형 세포. 100 밀리머 마그네슘 황산염과 5 밀리몰라 칼슘 염화 칼슘을 포함하는 신선한 LB의 300 마이크로 리터에서 상체를 버리고 세포 펠릿을 다시 중단하십시오.
다음으로, 미신심분리기 튜브에 기증자 파지 리자테의 100마이크로리터와 100마이크로리터를 결합하여 트랜스듀션 반응을 설정하였다. 이어서, 수신자 균주의 100마이크로리터와 LB의 100마이크로리터를 황산 마그네슘 및 염화칼슘과 결합하여 음극을 설정하였다. 인큐베이션 후, 구연산 나트륨 1개 200마이크로리터와 LB 1밀리리터를 두 튜브에 넣고 위아래로 부드럽게 배관하여 섞는다. 그런 다음 튜브가 한 시간 동안 배양 된 후 원심 분리를 통해 세포를 부드럽게 펠렛합니다.
원심 분리 후, 상체를 버리고 100 밀리머 나트륨 구연산나트륨으로 LB의 100 마이크로리터에서 펠렛 세포를 재연하십시오. 1X 암피실린을 장착한 LB 한천 플레이트에 변형된 전체 샘플을 용액과 파이펫을 피펫합니다. 마지막으로, 암피실린이 없는 LB 한천 판에 네거티브 대조군 세포 혼합물의 전체 부피를 피펫한다. 섭씨 37도에서 하룻밤 동안 플레이트를 배양한 후 멸균 파이펫 팁을 사용하여 트랜스듀션 플레이트에서 3~4개의 콜로니를 골라 1X 암피실린과 1개의 어금음 나트륨 100마이크로리터가 들어있는 새로운 LB 한천판에 적재합니다. 1개의 어금니 나트륨 구연산염의 단지 100 마이크로리터를 포함하는 다른 LB 화마 판에 음성 제어를 위한 이 도금 방법을 반복합니다. 그런 다음 파지가 없는 식민지가 성장할 수 있도록 밤새 섭씨 37도에서 플레이트를 배양합니다.
다음 날, 인큐베이터에서 접시를 제거하기 전에 70 % 에탄올로 벤치 상단을 닦아. 멸균 파이펫 팁을 사용하여, 트랜스듀션 플레이트에서 3개의 콜로니를 선택하고 LB 매체의 5밀리리터를 포함하는 별도의 튜브에 각각 추가합니다. 그런 다음, 음의 대조군 플레이트에서 3개의 콜로니를 선택하고 LB 매체의 5밀리리터를 포함하는 다른 튜브에 추가한다. 220 rpm에서 폭기와 흔들림과 함께 하룻밤 사이에 문화를 37도에서 성장시다. 이전에 설명한 대로 벤치 탑을 살균한 후 DNA 미니프렙 키트를 사용하여 제조업체의 지침에 따라 각 배양의 4.5 밀리리터에서 DNA를 분리합니다. 그런 다음, 35마이크로리터의 뉴클레아제 없는 물로 DNA를 엘테하고 실험실 분광계에 의한 결과 농도를 측정한다. 마지막으로, 글리세롤 의 0.5 밀리리터에 두 세균 성 솔루션의 나머지 0.5 밀리리터를 100% 글리세롤에 추가하여 글리세롤 주식을 준비하십시오.
트랜스듀션을 확인하려면 먼저 24qPCR 반응에 대해 두 개의 qPCR 마스터 믹스를 준비합니다. 첫 번째 마스터 믹스의 경우, 150 마이크로리터의 qPCR 버퍼 믹스를 마이크로센티르시페리지 튜브에 추가하고 12마이크로리터를 각각 암피실린 저항 유전자를 증폭하도록 설계된 전방 및 역 프라이머를 추가합니다. 다음으로, 마이크로센트심분리기 튜브에 150마이크로리터의 qPCR 마스터 믹스를 추가한 다음, 하우스키핑 유전자를 증폭하도록 설계된 12마이크로리터의 전방 프라이머와 역프라이머를 추가하여 두 번째 qPCR 마스터 믹스를 준비한다.
각 qPCR 반응에 대해 각 반응에서 100 마이크로그램의 실험 DNA를 qPCR 마스터 믹스의 14.5 마이크로리터와 결합합니다. 이제 이전에 설명한 대로 나머지 반응을 준비합니다. 예열된 온도 순환기로 반응을 섭씨 94도로 옮김한 다음 프로그램을 시작합니다. 마지막으로, qPCR에 의해 생성된 사이클 정량화 또는 Cq값을 사용하여 암피실린 저항 유전자의 정규화된 트랜스듀션 효율을 계산한다.
관심 있는 유전자에 대한 사이클 수량 또는 Cq값은 각각의 음의 대조군 및 변환된 샘플에 대해 표로 설정되었다. 이 예제에서 변환된 샘플과 같이 일반적으로 29사이클 미만의 낮은 Cq 값은 타겟 시퀀스의 높은 양을 나타냅니다.
또한 여기에 표로 된 가사 유전자는 각 반응에서 DNA의 양을 정상화하고 qPCR이 작동하는지 확인하기 위한 양성 대조군으로서 로딩 제어로 사용된다. 동일한 양의 가사 유전자가 적재되어 제공되고, 각 샘플에서 비교적 동일한 속도로 발견된다.
다음으로, 각 샘플에 대한 델타 Cq 값을 계산하기 위해, 해당 표적 유전자의 Cq 값으로부터 각 샘플에 대한 하우스키핑 유전자의 Cq 값을 빼낸다. 예를 들어 첫 번째 음수 컨트롤의 델타 Cq는 13.54입니다. 그런 다음 이 값을 사용하여 여기에 표시된 수식을 사용하여 각 샘플의 정규화된 변환 효율을 계산합니다. 마지막으로, 각 샘플 그룹에 대한 평균 정규화된 트랜스듀션 효율을 계산할 수 있다.
Subscription Required. Please recommend JoVE to your librarian.
Applications and Summary
박테리아에 의해 박테리아에서 유전자의 전송, 자연적인 과정 동안, 연구 목적의 무리에 대 한 매우 유용 입증. 변환 및 컨쥬게이션과 같은 유전자 전달의 다른 방법이 가능하지만, 트랜스듀션은 박테리오파지를 독특하게 사용합니다. 뿐만 아니라 숙주 게놈으로 유전자 통합을 허용, 뿐만 아니라 다른 방법에 취약 하지 않은 여러 박테리아에 유전자 전달에 대 한. 이 과정은 실험실에서 특히 유용하지만, 최근에 새롭게 떠오르는 유전자 치료 분야에서도 사용되어 왔으며, 보다 구체적으로는 대체 유전자 치료에서 박테리아를 활용하여 표적 조직에 치료제를 전달하는 치료 전략으로, 그 중 상당수는 다른 전달 방법에 취약하지 않고 많은 임상 적 관련성(8,9)을 가지고 있다.
Subscription Required. Please recommend JoVE to your librarian.
References
- Lederberg J, Lederberg E.M., Zinder, N.D., et al. Recombination analysis of bacterial heredity. Cold Spring Harbor symposia Quantitative Biol. 1951;16:413-43.
- Duckworth DH. "Who Discovered Bacteriophage?". Bacteriology Reviews. 1976;40:793-802.
- Yap ML, Rossman, M.G. Structure and Function of Bacteriophage T4. Future Microbiol. 2014;9:1319-27.
- Sulakvelidze A, Alavidze, Z., Morris, J. G. Bacteriophage Therapy Antimicrobial Agents and Chemotherapy 2001;45(3):649-59.
- Moore S. Sauer:P1vir phage transduction 2010 [Available from: https://openwetware.org/wiki/Sauer:P1vir_phage_transduction].
- Kobayashi A, et al. Growth Phase-Dependent Expression of Drug Exporters in
- Escherichia coli and Its Contribution to Drug Tolerance. Journal of Bacteriology. 2006;188(16):5693-703.
- Rocha D, Santos, CS, Pacheco LG. Bacterial reference genes for gene expression studies by RT-qPCR: survey and analysis. Antonie Van Leeuwenhoek. 2015;108:685-93.
- Pálffy R. et al. Bacteria in gene therapy: bactofection versus alternative gene therapy. Gene Ther. 2006 13:101-5.
- O'Neill JM, et al. Intestinal delivery of non-viral gene therapeutics: physiological barriers and preclinical models. Drug Discovery Today. 2011;16:203-2018.