Source: Susannah C. Shissler1, Tonya J. Webb1
1 Département de microbiologie et d’immunologie, Université du Maryland, Baltimore, MD 21201
L’immunoprécipitation (IP, également connue sous le nom d’un «pull-down» d’essais) est une technique largement utilisée qui a des applications dans une variété de domaines. Conçu pour la première fois en 1984, il a été affiné en 1988 (1, 2). L’objectif fondamental de la propriété intellectuelle est la purification et l’isolement d’une protéine spécifique à l’aide d’un anticorps contre cette protéine. Le mot «immuno» fait référence à l’utilisation d’un anticorps tandis que le mot «précipitation» fait référence à l’arrêt d’une substance spécifique d’une solution. La protéine cible peut être endogène ou recombinante. La plupart des protéines recombinantes ont une étiquette d’épitope (c.-à-d. myc ou drapeau) attachée à elles pour simplifier la purification suivante. Typiquement, il est plus facile d’optimiser la protéine recombinante IP parce que les anticorps contre les étiquettes d’épitope recombinant sont très forts et efficaces. Les anticorps contre les protéines endogènes ont une efficacité extrêmement variable – ce qui rend beaucoup plus difficile d’optimiser ces adresses IP. Une étape nécessaire après l’immunoprécipitation est la vérification de la purification. La protéine isolée est résolue à l’aide de SDS-PAGE et par la suite sondée pour la pureté par les taches occidentales (figure 1). Un contrôle important est l’utilisation d’un anticorps différent pendant la tache occidentale pour vérifier tirer vers le bas de la protéine correcte. La combinaison de la propriété intellectuelle avec les techniques suivantes est un outil d’analyse puissant. L’objectif après la purification peut être la caractérisation de la protéine elle-même par la RMN, la spectrométrie de masse et les essais in vitro, ou l’analyse des partenaires interagissant de la protéine (c.-à-d. protéine, ADN, ARN) (3, 4, 5).
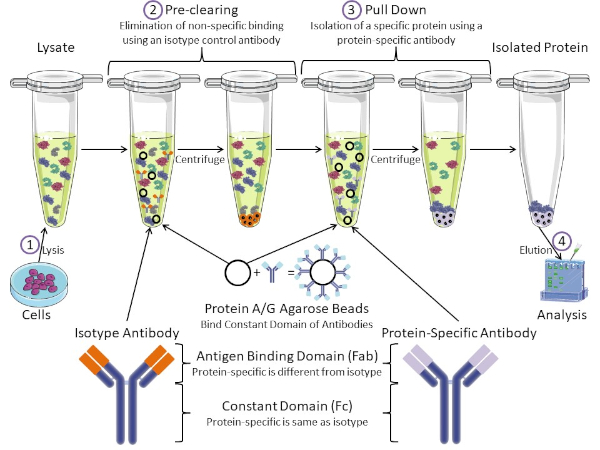
Figure 1 : Aperçu de la procédure d’immunoprécipitation. L’immunoprécipitation est l’isolement d’une protéine spécifique à l’aide d’un anticorps. Après la production de lysate à partir de cellules, il y a deux étapes principales- pré-dédouanement et tirer vers le bas. Pendant l’étape de pré-dédouanement, les lysates cellulaires sont pré-dédouanés des protéines qui se lient aux anticorps non spécifiquement à l’aide d’un anticorps anti-isotype. Dans l’étape de traction vers le bas, la protéine cible est tirée vers le bas à l’aide d’un anticorps protéique spécifique. La protéine isolée est ensuite analysée par Western blot. Les anticorps isotypes et les anticorps protéiques spécifiques ont le même domaine constant, mais différents domaines de liaison d’antigène. Un composant clé de ce protocole est protein A/G perles d’agarose qui lient le domaine constant des anticorps- permettant l’immunoprécipitation de la protéine cible. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
Les anticorps sont la composante clé d’une immunoprécipitation qui la différencie des autres formes de purification des protéines (c.-à-d. la purification de la colonne d’affinité au nickel). Les anticorps sont des molécules fabriquées par des cellules B qui peuvent reconnaître des épitopes protéiques spécifiques. Les anticorps ont deux domaines : constante (Fc) et liaison antigène (Fab) (figure 1). Le domaine constant identifie le type d’anticorps et dicte la fonction in vivo. Habituellement, les domaines constants des anticorps utilisés pour la propriété intellectuelle sont la souris, le rat ou le lapin IgG. La partie de liaison d’antigène de l’anticorps reconnaît un épitope spécifique d’une protéine spécifique. Les anticorps peuvent reconnaître les épitopes sur les protéines pliées qui peuvent ne pas exister lorsque la protéine est dénaturée et vice versa. Par conséquent, la disponibilité de l’épitope dépend du pliage des protéines – identifier un facteur important à considérer lors du choix des anticorps et des conditions pour la propriété intellectuelle.
Les systèmes procaryotes et eucaryotes ont des protéines liant les anticorps. Dans les systèmes eucaryotes, le but est la protection immunitaire contre les bactéries tandis que dans les systèmes procaryotes, le but est la protection contre le système immunitaire. Les protéines liant les anticorps affectent la méthodologie de la propriété intellectuelle de deux façons. Tout d’abord, il y a une étape nécessaire de pré-dédouanement (figure 1) pour débarrasser le lysate des protéines qui lient les anticorps – réduisant ainsi la liaison non spécifique dans le produit final. Cette étape utilise un anticorps isotype qui a le même domaine constant que, mais un domaine de liaison d’anticorps différent de votre anticorps spécifique aux protéines. Les protéines bactériennes liant les anticorps sont le deuxième composant clé de cette méthode. Après que l’anticorps protéique-spécifique lie la protéine cible, l’anticorps : complexe de protéine doit être tiré vers le bas (figure 1). Les protéines A, G et L sont des protéines bactériennes qui lient le domaine constant des anticorps. Alors que les bactéries l’utilisent pour subvertir le système immunitaire, les chercheurs ont coopté ce système pour la purification facile des anticorps, et il est utilisé à la fois pendant les étapes de pré-dédouanement et de traction. Ces protéines ont des affinités de liaison différentes pour différentes espèces et différents sous-types de domaine constants – un autre facteur à considérer lors du choix des conditions pour la propriété intellectuelle. De nombreuses entreprises vendent des perles d’agarose étiquetées Protéines A/G (figure 1), des colonnes de spin préfabriquées ou des résines pour fabriquer des colonnes. En général, les perles et les colonnes de spin sont utilisées pour de plus petites tailles d’échantillon tandis que les résines sont utilisées pour la purification en vrac.
Dans cet exercice de laboratoire, nous démontrons comment purifier la protéine endogène c-myc, des thymocytes murines primaires, utilisant la protéine A/G Plus agarose perles basées sur la technique d’immunoprécipitation de base. Le protocole commence à partir de la préparation du lysate cellulaire et se termine par la vérification de la protéine réussie tirer vers le bas en utilisant l’analyse de tache occidentale.
The results of the procedure detailed above are shown in Figure 2. From left to right, the lanes contain the control group (isotype), the test group (c-myc), the pre-cleared lysate (lysate), and the molecular weight ladder (ladder). The 25 and 75 kDa ladder bands are marked. The two prominent bands at ~25 kDa and 50 kDa are the light and heavy chain of the binding antibody, respectively and are non-specific to the IP or the samples. c-myc protein which runs around 67kDa on Western blots and is usually visible just below the 75 kDa ladder band. In this blot, the c-myc band is visible in the second lane, but absent in the first lane, indicating that the IP antibody successfully pulled down c-myc. There is no visible band in the pre-cleared lysate lane, suggesting that this protein has low endogenous expression levels.

Figure 2: Results of a Western Blot Analysis, used to assess the purification of c-myc by immunoprecipitation. A band at 67 kDa, corresponding to c-myc, is visible in the anti-c-myc lane, but not the isotype control lane. Note that c-myc levels were not high enough to be visualized in the lysate lane. Please click here to view a larger version of this figure.