To date, immunostaining in live tissue is not typically used due to formation of immune complexes leading to high staining background and immunotoxicity20. This was overcome by pre-blocking Fcγ receptors on tissue macrophages, thus “blinding” these cells to subsequent indirect strong immunostaining. As the labeling was extracellular, phototoxicity and fluorophore bleaching could be controlled by immersion of the tissue in natural antioxidant buffered, isosmotic ascorbic acid (Fig. 1A). Since our initial description of this method5, we improved our anti-bleaching and anti-phototoxic technique so that photobleaching can now not only be inhibited but completely prevented (Fig. 1B). This is done by embedding the ear in a large volume of antioxidant (100 μl) within the chamber where the ear is immobilized. Note that 300 seconds constant imaging time corresponds to 10 hours of imaging when pictures are collected for 500 msec every one minute (the average imaging settings).

Figure 1. Photobleaching and phototoxicity was stopped by replacing chloride with ascorbate in Ringer’s buffer and embedding the exposed ear in 100 μl volume of that solution. (A) Tissue was stained first with a biotinylated antibody against collagen IV, the component of the basement membrane. The staining was later detected with streptavidin-647 (red), and then constantly imaged for 300 seconds in either normal Ringer’s buffer (upper panel) or ascorbate-Ringer’s (lower panel). Note that 300 seconds constant imaging time corresponds to 10 hours of imaging when pictures are collected for 500 msec every one minute (the usual imaging settings). The brightest 25% of pixel intensity values are green colored. (B) Quantification of immunofluorescence decay (50% in Ringer’s vs. 0% in ascorbate-Ringer’s). Values were normalized to the initial fluorescence. Images collected with immunofluorescence stereomicroscope. Scale bar in A, 100 µm. Please click here to view a larger version of this figure.
Investigating the interaction between tumor metastatic cells and tumor stroma is crucial to understand the process of tumor cell migration and tumor immunity. This is because the tumor-associated stroma composes up to 90% of the tumor mass and actively regulates tumor growth and metastatic spread21. However, mechanistic insight into how the matrix drives tumor cell migration towards either lymphatic or blood vessels is lacking 22. This is partially due to the fact that intravital imaging of matrix proteins is limited to the visualization of matured fibers of fibrillar collagens using second harmonic generation in two-photon microscopy23. Therefore, we adapted our method for the visualization of the tissue microenvironment including blood and lymphatic vessels, pericytes, nerves, muscle and adipocytes to include the direct visualization of extracellular matrix components. Many structures can be distinguished based on their morphology after immunostaining for basement membrane components such as collagen IV or perlecan (Fig. 2A), however, direct staining for specific cell surface markers, e.g. Lyve1 (Fig. 2B and C) and podoplanin (Fig. 2C), further allows distinguishing initial, capillary lymphatics (Lyve1+) from lymphatic collectors (Lyve1–). Staining for matrix-bound CCL21 in the skin revealed deposits of this chemokine on the basement membrane of lymphatic collectors identified with the staining for perlecan, the heparan sulphate proteoglycan (Fig. 2D).
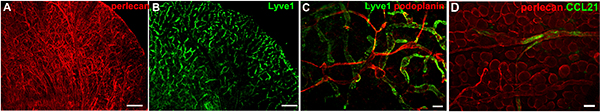
Figure 2. Examples of live staining in a normal mouse ears. (A) Staining for perlecan, a basement membrane component, depicts all blood and lymphatic vessels, nerves, muscle fibers and adipocytes. (B) Lyve1 staining marks the initial lymphatic capillary network. (C) Co-staining for Lyve1 and podoplanin, a pan-lymphatic marker, depicts networks of initial and collecting lymphatics. The dorsal ear dermis can be imaged using classic epifluorescence microscopy (without optical sectioning) as the ear dermis has low number of adipocytes that would otherwise cover the imaging field. (D) CCL21 staining (green) on lymphatic basement membrane stained for perlecan (red). Images collected with immunofluorescence stereomicroscope. Scale bars in A and B, 1 mm; in C, 100 μm and 50 μm in D. Please click here to view a larger version of this figure.
Most importantly, structural proteins that normally cannot be detected by SHG, the classic matrix detection method23, can be visualized. For example, we found that tenascin C (Fig. 3A), a matrix protein that is expressed during tumorigenesis, wound healing and inflammation19 is deposited in different locations of tumor stroma than fibrillar collagens (Fig. 3B). This matrix heterogeneity might affect tumor cell distribution and metastasis (Fig. 3C).

Figure 3. The dorsal ear dermis can be live imaged using multi-photon microscopy. A single field of tumor B16-F10. Tumor stroma was labeled with tenascin C antibody and the staining was detected with anti-goat-594 donkey antibody. Immunofluorescence of tenascin C (red) network is shown in A and fibrillar collagens detected with second harmonic generation (SGH) in B. These two networks are superimposed with tumor cells (cyan) in C (merged). New tumor matrix marked by tenascin C is not overlapping with fibrillar collagens detected with SHG (green). The image with 44, four times averaged z-sections with z-step 1.0 μm was acquired in 16 bit color depth mode. Images collected with two-photon microscope. Scale bar, 100 μm. Please click here to view a larger version of this figure.
Following the immunolabeling of different tumor matrix components (e.g. tenascin C and collagen IV) of tumor matrix we could identified restricted and sudden events of tumor matrix directional remodeling (Video 1). Forces that develop within tumor microenvironment may lead to expansion or contraction of the tumor matrix and in consequence remodeling and elongation of the tumor vasculature to a similar extent as we showed previously in the case of healing wounds11,24.
Video 1. Expansion of tumor matrix labeled with collagen IV (red) and tenascin C (cyan) elongate blood vessels (arrow) and passively translocate three tumor cells (green). Localized contraction of teascin C-rich tumor matrix translocate blood vessel (red horizontally oriented structure) by approximately 100 μm during 12 hours of imaging. Images collected with immunofluorescence stereomicroscope. Click here to view video.
Using multi-photon microscopy with fluorophore labeling is problematic as photon flux used in these experiments will quickly bleach fluorescent dyes that are not protected from oxidation25. Here we show that we can perform two-photon time-lapse microscopy while simultaneously imaging immunolabeled tenascin C matrix with minimal photobleaching of fluorophores even in high photon density two-photon microscopy (Video 2).
Video 2. Low level photobleaching of tenascin C immunofluorescence (red) during two-photon microscopy. B16-F10-GFP tumor cells (cyan) were imaged in the open dorsal ear 9 days after inoculation. The fluorescent signal was protected by application of ascorbate-Ringer’s buffer onto the imaged tissue. Second harmonic generation (green) is not overlapping with new matrix represented by tenascin C. 16 bit color depth image with 11, four times averaged z-sections with z-step 1.9 μm was acquired for 15 min. Images were collected every 1 minute 5 seconds in 6 z-planes collected. Click here to view video.
We also imaged immune-cell interactions with metastatic tumor cells. CMTPX-labeled splenocytes from a tumor-bearing mouse extravasated from tumor-associated blood vessels 8 hours after i.v. transfusion, invaded the tumor matrix and actively interacted with tumor cells by forming long-lasting cell contacts (Video 2). Fluorophore-647-labeled collagen IV structures were resistant to photobleaching during the 12 hours of imaging.
Video 3. Interaction of CMTPX-labeled splenocytes from tumor (B16-F10)-bearing mouse with individual tumor cells (GFP- B16-F10). Splenocytes were i.v. transfused after tissue staining for collagen IV. Collagen IV-green (647), splenocytes-red (CMTPX), B16-F10 cyan (GFP). Images collected with immunofluorescence stereomicroscope. Video duration 12 hours. Click here to view video.
Freshly isolated tumor cells were overlaid on the exposed ear dermis pre-stained for collagen IV. We could observed that after adhering to the tissue some groups of cells started collective migration along basement membrane of blood vessels and adipocytes.
Video 4. Tumor cells migrate along basement membranes. Normal ear skin stained for collagen IV (647, red) was overlaid with freshly passaged B16-F10-GFP cells and imaged for 5 hours. Some tumor cells that adhered to tissue started collective migration along basement membrane of blood vessels and adipocytes. Images collected with immunofluorescence stereomicroscope. Video duration: 5 hours. Click here to view video.
Also, because the ear dermis is virtually two-dimensional, we could readily collect large volumes of data using fast fluorescence stereomicroscopy, instead of slower and more expensive confocal or multiphoton microscopy. However, it is also possible to use any of the mentioned scanning microscopy methods with our intravital IF preparations (Fig. 3, Video 1).
