Neuronal tissue is highly complex; a heterogeneous cellular mixture that is spatially ordered within defined layers and compartments and with plastic connectivity via cell contacts and especially via axon and dendrite outgrowths. New techniques are required to confer greater experimental freedom to gain deeper insights and unravel mechanisms central to disease, development, and healthy function. The Campenot chamber1,2 and more recently microfabricated embodiments3,4 can be used for the ex vivo preparation of networked neuronal co-cultures with the ability to selectively perturb the different somatic populations and also their neurite outgrowths. These microfluidic devices have for example been used to study axon degeneration and regeneration following chemical5,6 or laser axotomy6-8, tauopathy9, viral dissemination10,11, and mRNA localization in axons4.
To extend the reach of the neurobiologists, technological developments are required to prepare minimalistic neuronal co-cultures. This enables disentanglement of the neuronal network for the investigation of the system with single cell and sub-cellular precision. The requirement for minimal cell numbers opens the possibility to analyze rare cell types, including dopaminergic substantia nigra cells relevant to Parkinson's disease, spiral ganglia from the ear, peripheral neurons, and stem cells. Beyond this, cellular economy is relevant to the 3Rs initiative. Using these microfluidic platforms, large-scale toxicity screens or other high throughput, data rich experimental series requiring animal neurons can now be considered.
In this paper protocols for the fabrication and use of a microfluidic device are described. Microfluidic arraying in combination with an in situ biomaterial patterning method can be used for the registration of highly interconnected neuronal co-cultures using minimal cell numbers. Microfluidic arraying is based on a differential flow approach12–15, whereby microstructured traps are positioned within a fluidic circuit (illustrated along with SEM images in Figure 1). The path 0 → 1 has the lower fluidic resistance (R2 > R1) for transporting neurons to a linear array of microstructured apertures – the inlets to the neurite outgrowth channels. Occupancy of the trap by a single cell locally impedes the flow to divert the streamlines for trapping subsequent cells in neighboring traps. Complete occupancy of the traps in the array switches the fluidic ratio (R1 > R2) to divert the streamlines into the serpentine path (0 → 2) to produce a bypass mode of operation for the removal of excess neurons.
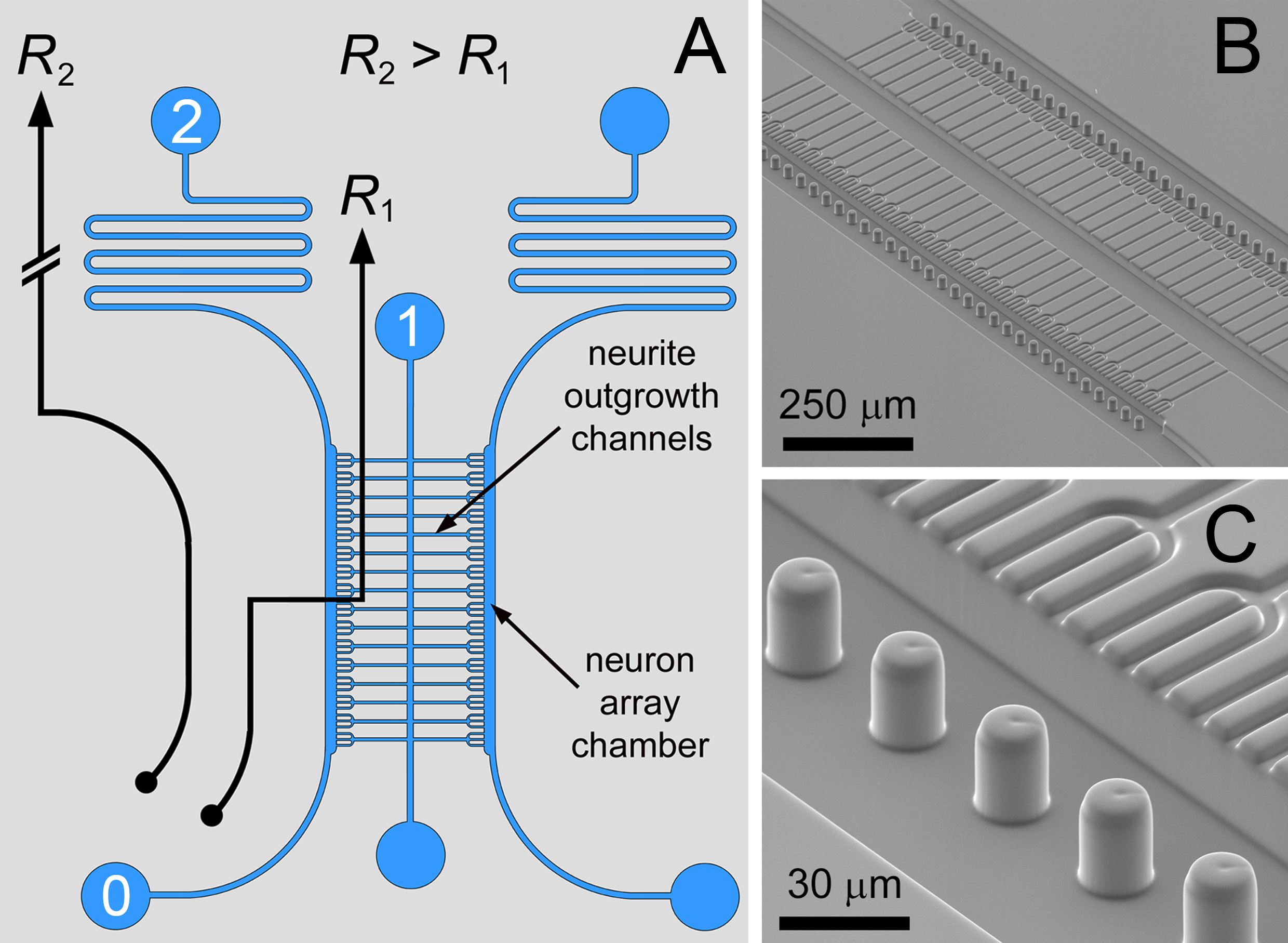
Figure 1. Microfluidic Circuit. A) The differential resistance fluidic circuit for single neuron arraying, with flanking culture chambers interconnected by neurite outgrowth channels. B,C) SEM images of the bilayer compartmentalized neuron co-culture array with meniscus pinning micropillars. With this design, trident-shaped neuron trapping structures were used to promote fasciculation of the neurite outgrowths. Figure and legend reproduced with permission of the Royal Society of Chemistry (RSC)12. Click here to view larger image.
The preparation of micropatterned neuronal networks on planar substrates can readily be achieved (for examples from our group, see Frimat et al.16 and Heike et al.17). However, encapsulating bioactive material patterns within PDMS devices and with the requirement of micrometer-scale alignment of these to the microfluidic channels poses a major technical challenge. In section 3.1 a protocol for the in-chip, or in situ, preparation of biomaterial patterns is presented. These patterns enable neuron registration during lengthy culture timescales and promote outgrowths between compartments. Meniscus-pinning microstructures are used to align a so-called water mask with the neuron arraying sites and neurite outgrowth channels. The water mask protects adhesion molecule coatings during plasma treatment, whereas exposed surfaces are disintegrated to define the biomaterial pattern. In addition, protocols are provided for cell culture and for fluidic isolation necessary for the selective treatment of the different co-culture compartments.
The protocols are designed to leverage the user-friendly principles of soft lithography for the replication of poly(dimethylsiloxane) (PDMS) microfluidic devices18. Similarly, in situ biomaterial patterning is straightforward, exploiting evaporation and surface tension phenomena, and only requiring an inexpensive hand-held plasma source. The microfluidic circuit effectively programs cell loading and compartment specific treatments making these operations simply a matter of dispensing materials into the correct bottom port and aspirating from above. In this manner, it is intended to give the neurobiologists the freedom to prepare and use microfluidic devices in their own laboratories.
The protocols are intended for neuroscientists, and are summarized in Figure 2. As such it is recommend that microfabrication of the SU-8 masters used for PDMS replication is outsourced to commercial or institutional facilities. The two layer mask designs are freely available as supplementary information (ZIP) on the Royal Society of Chemistry website12. Importantly, the first SU-8 layer should be fabricated to a depth of 2.5-3.0 μm, and the second to a depth of 25-30 μm. These are key dimensions necessary for effective neuron arraying. The 3 micron height limit is also necessary to prevent neurons being transported or migrating between the two compartments12.
1. Microfluidic Device Replication in PDMS
- Mix the PDMS prepolymer thoroughly with the curing agent (184) at a ratio of 10:1 and degas the mixture in a vacuum desiccator for typically 20 min. Alternatively, put the mixture in a 50 ml Falcon tube and use low g centrifugation for degassing.
- Place the 2-layer microstructured SU-8 wafer on an 80 °C hotplate and align polymer frames to each device on the wafer. Pour PDMS into the frames to a depth of ≥5 mm and allow >1 hr to ensure complete thermal curing.
- Once cured, remove the wafer from the hotplate and allow to cool on a flat surface.
- Wear protective eyewear and work from one corner (away from the SU-8 microstructures) with a rigid scalpel to gently prize the frame and PDMS from the wafer.
- Remove the PDMS mold from the frame and use a pair of scissors to trim the device of excess PDMS. This so-called flashing is often produced when a thin film of PDMS spreads between the polymer frame and the wafer surface.
- Remaining PDMS can be applied to the entire wafer and cured to protect the wafer during storage. Remove this prior to subsequent device molding, and in doing so remove any particulate contaminants from the surface.
2. Device Assembly and Interconnection
- Prepare the 6 interface ports using a 3 mm diameter biopsy punch. Work on a dark surface, in good light conditions and with the microstructured features facing upwards to aid alignment of the ports with the microfluidic channels.
- Inspect the device for particulates. Remove these using reversible adhesive tape.
- The microfluidic circuit is encapsulated using a thin PDMS layer mounted on a glass coverslip: These are prepared by pouring a small volume (~0.5 ml) of the PDMS-curing agent mixture (10:1, 601) onto the base of a flexible, bacteriological grade Petri dish. Gently press a coverslip onto the PDMS layer, place on the hotplate and thermally cure for a few minutes. Quickly after cooling, use a scalpel to cut out and prize the coverslip-PDMS bilayer from the Petri dish. Trim with scissors if necessary.
- Plasma bond the PDMS device and support layer to produce a good seal: Optimum conditions are instrument specific. For example, a plasma oven operating at 70 W and 40 kHz in a 0.2 mbar oxygen atmosphere for 40 sec produces an excellent PDMS–PDMS bond. Once plasma treated, press both parts together and leave for a minute to fully bond.
- Use silicone tubing (1.6 mm ID; 3.2 mm OD) to interface with pumps and syringes.
3. Biomaterial Patterning and Microfluidic Operations
The protocols for in situ biomaterial patterning, neuron culture and fluidic treatments are illustrated in Figure 2.

Figure 2. Illustrated microfluidic protocols. A) PL and PLL-g-PEG patterning; addition of the cell adhesion layer (e.g., PL, green), with evaporation producing the aligned water mask. B) Atmospheric air plasma treatment of the exposed PL; two antenna pins are used, with the plasma introduction antenna illustrated with a pink star. C) PBS (blue) washing by aspiration is used to avoid contaminating the plasma-treated region with PL. D) Delivery of PLL-g-PEG (red) by aspiration to coat the plasma-treated regions. E) Cell loading; simultaneous microfluidic arraying of neurons to both flanking compartments by aspiration, F) media (pink) perfusion using a hydrostatic-driven flow. G) Isolated fluidic treatments; peripheral treatment of a test agent (black) delivered from the bottom flanking inlet by aspiration and H) central treatment delivered from the bottom central inlet by aspiration. Figure and legend reproduced with permission of the Royal Society of Chemistry (RSC)12. Click here to view larger image.
Immediately following plasma bonding the PDMS microfluidic channels are highly hydrophilic (contact angle ≤5°). Such surfaces are suitable for the adhesion and culture of cell lines such as differentiated human SH-SY5Y neuron-like cells. For the ex vivo culture of primary neurons and the culture of neuronal precursor cell lines such as the Lund human mesencephalic cell line (LUHMES) a polyamine coating, either polylysine (PL) or polyornithine (PO), is required as an electrostatic anchor. In addition adhesion proteins such as laminin or fibronectin are often required as the integrin-interfacing coating. However, the PL and PO coatings readily bind neurons, hindering the microfluidic delivery to the array sites and also imposing unwanted shear stresses. To solve this problem in-chip biomaterial patterning is required to localize adhesion materials at the trap sites and the outgrowth microchannels. This also promotes the development of inter-compartment connections and reduces local entanglement. The protocol also involves the addition of PEG materials to neighboring areas to ensure proteins and neurons are restricted to the desired locations during lengthy culture. The following protocol is documented in Figure 3:
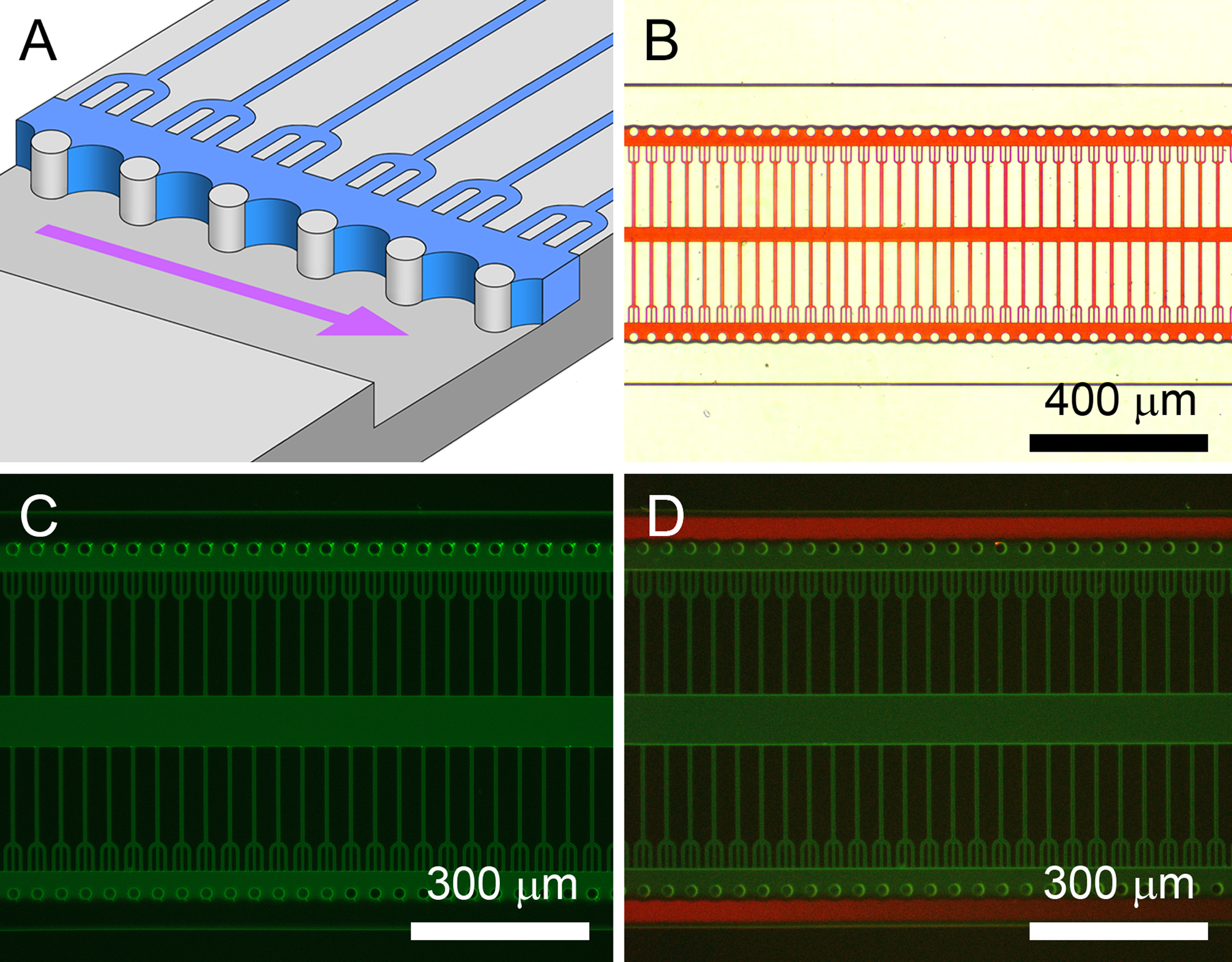
Figure 3. Water masking for biomaterial patterning by plasma stenciling. A) Illustration of the water masking process with the PDMS micropillars pinning the water meniscus in place for plasma stenciling. Curvature of the meniscus in the vertical plane has been neglected. B) Water mask visualized with a red dye and positioned by meniscus pinning. C) The patterned PL-FITC coating imaged after plasma stenciling. D) Addition of protein rejecting and cell repellent PLL-g-PEG–TRITC to a plasma-patterned PL-FITC coating. This was used for the registration of SH-SY5Y cells and also for patterning an additional fibronectin layer for LUHMES cells. Figure and legend reproduced with permission of the Royal Society of Chemistry (RSC)12. Click here to view larger image.
- Water Masking for In situ Biomaterial Patterning by Plasma Stenciling
- Shortly after plasma bonding pipette a 0.5 μl volume of PL or PO (100 μg/ml in 1x PBS) into the bottom center port. Within a matter of seconds the entire microfluidic circuit is filled by capillary action. Leave for a few minutes to fully coat the hydrophilic PDMS surfaces with the polyamines.
- Remove unbound polyamine molecules by aspiration-driven washing with 1x PBS (see Figure 2C) for 1 min.
- Remove all PBS from the ports and heat the device by illumination with a microscope. This increases evaporation from the ports for rapid water mask formation. The water mask (illustrated in Figure 3A and documented in Figure 3B) is established within ~5 min.
- Inspect the device by microscopy to ensure water masking is complete. Please note reservoir structures upstream of the array site are included in the mask design to prolong the stability of the water mask (~15 min).
- Insert stainless steel pins into the ports (Figure 2B). These are used to couple the plasma generated by an atmospheric pressure hand-held corona discharge or Tesla generator (e.g., operating at 2 MHz, 30 kV signal)19,20 into the vacant microfluidic channels. Point the tip of the plasma source close to the tip of one pin and plasma treat the microchannel for ≤1 sec. Use low light conditions to observe plasma patterning.
- Remove the water mask with a wash step. Use parallel aspiration from the upper 3 ports. Use 4 equal lengths of silicone tubing connected by a 4-way adaptor to an aspiration pump to achieve parallel aspiration. Start aspiration and dispense PBS into the bottom 3 ports to draw PBS through the microfluidic circuit. In this manner a PDMS-polyamine material contrast is produced (see Figure 3C). Evacuate PBS from the microfluidic circuit and leave for several hours to restore the native, hydrophobic state. This renders the PDMS fully non-permissive to cell adhesion.
- Should additional adhesion materials, such as laminin or fibronectin, be needed the following steps are required and should be done immediately after removing the water mask: Pipette a 10 μl volume of the graft co-polymer poly-L-lysine–poly(ethylene glycol) (PLL-g-PEG, 100 μg/ml, in PBS) into both bottom flanking channels, followed by aspiration from the upper flanking channels for 2 min.
- Without interrupting the flow, remove excess PLL-g-PEG by exchange with a PBS buffer. This selectively coats the non-polyamine-coated regions with the PEG moiety acting to prevent protein adsorption and cell adhesion17,21.
- Adhesion proteins such as laminin or fibronectin can then be co-localized with the poly-amine coating: Pipette 10 μl volumes of these ECM proteins (typically 10 μg/ml) into all three bottom ports. Aspirate from the top three ports until the device is filled with the protein solution and incubate for 1 hr to produce a good quality coating.
- Remove excess proteins by aspirating PBS from the bottom 3 ports.
- Microfluidic Single Neuron Arraying
- Prime the devices with appropriate media. Add 20 μl volumes to the bottom 3 ports and by parallel aspiration from the upper three ports rapidly fill the microfluidic circuit with media. Use a 4-way tubing connector to couple the 3 upper ports via equal-length silicone tubing to an aspiration pump.
- Add 20 μl of a disaggregated cell suspension (1 x 106 cells/ml) to each of the flanking inlet ports (0, in Figure 1).
- Equip an aspiration pump with a flow regulator or use a syringe for gentle aspiration from the upper 3 ports. Complete neuron arraying typically requires 1 min.
- Harvest excess neurons remaining in the 4 flanking ports with a pipette for arraying in subsequent microfluidic devices.
- Remove the tubing. Fill all ports with media and place the device in the incubator. Within a few hours the cells become fully adherent on the biomaterial pattern.
- Neuron Culture, Selective Fluidic Treatment, and Immunostaining
- Exchange the media by periodic perfusion using hydrostatic feed (a 2 mm column height difference is appropriately slow).
- Alternatively, submerge the microfluidic device in media, exchanging the media periodically (2–3 days) as with standard cell cultures.
- Selective fluidic treatments to either neuron culture compartment or the central neurite outgrowth compartment are achieved as follows: Dispense 20 μl of the test substance into the bottom port of the channel of interest, and aspirate from the port directly above. For lengthy treatments reduce the flow rate using a flow regulator and replenish the test substance as required.
- Immunostaining protocols vary for each molecular target and associated reagents. In order to counter the potential risk of shear-induced damage to the neurons a slow flow rate was used. This results in increased application times to fully perfuse the system, extending normal protocol times. Hydrostatic perfusion using ports with differing column heights (e.g. ,by 2 mm) was used to deliver reagents. More rapid reagent addition and removal using a pump can be used to reduce experimental timescales. Connect an aspiration pump to the upper 3 ports to draw reagents from the bottom 3 ports.
Water masking exploits surface tension. The pressure tolerance at the air-liquid interface scales inversely with the radius of curvature [ , producing highly stable interfaces at microfluidic dimensions. The micropillars effectively anchor the water mask in place. Water mask formation is driven by evaporation from the microfluidic ports, with the rate increased by heating. Using the heat from low magnification (4X – 10X) microscopy illumination, the water mask was typically establishing in ≤5 min. Evaporation also leads to the eventual collapse of the water mask. To increase the lifetime of the water mask to ~15 min (practical for plasma stenciling), 18 nl reservoir structures have been positioned upstream of the culture compartments. The rate of water mask formation and collapse can vary, and intermittent observation is recommended to identify a suitable period for plasma treatment. Nevertheless, multiple devices can be prepared simultaneously.
, producing highly stable interfaces at microfluidic dimensions. The micropillars effectively anchor the water mask in place. Water mask formation is driven by evaporation from the microfluidic ports, with the rate increased by heating. Using the heat from low magnification (4X – 10X) microscopy illumination, the water mask was typically establishing in ≤5 min. Evaporation also leads to the eventual collapse of the water mask. To increase the lifetime of the water mask to ~15 min (practical for plasma stenciling), 18 nl reservoir structures have been positioned upstream of the culture compartments. The rate of water mask formation and collapse can vary, and intermittent observation is recommended to identify a suitable period for plasma treatment. Nevertheless, multiple devices can be prepared simultaneously.
Plasma stenciling requires ~1 sec. The use of lengthy treatments vaporizes the water mask, leading to excessive plasma disintegration of the polyamine coating. On occasion insufficient plasma treatment results, leaving intact polyamine coatings on the adjacent microchannel wall (see Figure 3D) that can support neuron adhesion12. Nevertheless, arrayed mouse cortical neurons had a patterning efficiency of 89.5%12 following culture for 6 days. This efficiency is equivalent to PL/PEG patterns on planar substrates. Biomaterial patterning has the further merit of promoting outgrowths into the neurite microchannels. With biomaterial patterning outgrowths extended within ~90% of the microchannels, whereas with a continuous polyamine coating outgrowth levels were reduced to ~70%. Using cortical neurons, 7 days of in-chip culture was required for the outgrowths to span both compartments (a distance of 500 μm). Differentiated human SH-SY5Y neurons had higher outgrowth rates, establishing connections in 4-6 days. Cortical, LUHMES and SH-SY5Y neurons have been cultured for up to 2 weeks. This is sufficiently long for spontaneous electrophysiological function to be attained and can be indirectly measured by Ca2+ imaging. This would validate the widespread applicability of the biomaterial patterning and microfluidic arraying methods for establishing functional neuronal networks.
The meniscus-pinning structures do, however, promote neurite entanglement (see Figure 4C). Edge finding is a common feature of axons cultured on microstructured surfaces22. To prevent such entanglement and further promote the formation of inter-compartment connections, meniscus-pinning pillars can be merged with the microchannel wall (i.e. no longer free-standing). In addition, outgrowths in the central microfluidic channel can be disorganized. For experiments requiring linear connectivity, the central channel can be removed (see the mask designs in Dinh et al.12, supplementary information). This circuit requires serial arraying of the first compartment, followed by a period (e.g., 4 hr) for cell adhesion or so-called cellular valving14 to render the circuit permissive for arraying cells in the second compartment.
The microfluidic circuit provides pre-programmed fluidic operations. The different functions are determined by the choice of port to load reagents or interface with tubing or tips. This avoids the need for complex, costly and space demanding feedback-controlled instrumentation to make the technique highly accessible to the neurobiologists. Circuit design is critically dependent on optimal relative dimensions for achieving microfluidic arraying12-14. Small changes can prevent the circuit from functioning, and we therefore recommended initial adopters of the protocols to make use of the freely available designs12. With an optimal circuit, microfluidic single neuron arraying is rapid, with ~100 neurons arrayed in each culture chamber in less than 1 min. Despite the limited number of neurons, the local density is still equivalent to standard cultures to provide sufficient life-support signaling levels. As documented in Figure 4B this approach effectively yields 1 neuron per trap (1.14 ±0.09)12. However, not all neurons are arrayed. The microfluidic circuit generates parallel streamlines, such that neurons arriving in the outer regions of the laminar flow continue to the serpentine channel. Nevertheless the presented system is 10-100 fold more economical with cells than existing compartmentalized systems. Expelled cells can be harvested from the ports and tubing. In future designs, upstream deterministic lateral displacement structures23 can be used to deflect all cells into the arraying streamlines for absolute cellular economy.

Figure 4. Microfluidic neuron arraying. A) Manual aspiration using a syringe is used for gentle neuron arraying. A 4-way tubing connector is used to interface the syringe with the upper 3 ports for simultaneous neuron arraying in both compartments. B) Differentiated neuron-like human SH-SY5Y cells and other neurons are arrayed with single cell precision (one neuron per trap). C) Following 5 days of culture, neurite extensions interconnect the two cultures. Nuclei are stained with DAPI and actin immunostaining was used to visualize the outgrowths. Modified figure and legend reproduced with permission of the Royal Society of Chemistry (RSC)12. Click here to view larger image.
A key feature of compartmentalized systems is the ability to selectively treat individual cell populations to investigate material trafficking and long-range signaling. However, methods available in the literature are either inadequate for fluidic isolation or are poorly described. The fluidic isolation method we have developed enables the rapid delivery (<1 min) of test substances and prolonged fluidic isolation (>1 hr)12. Results are documented in Figure 5. This capability is made possible by the combination of the circuit design and by using aspiration to deliver the test substance. This is in contrast to fluid injection using either a syringe pump of by hydrostatic feed. Despite the high fluidic resistance of the neurite outgrowth channels, these methods produce convection paths that rapidly disperse test substances and contaminate the entire device. Aspiration from the upper port also interacts with fluid throughout the circuit (a feature central to the differential resistance arraying principle). However, in this mode of operation, media are drawn from elsewhere in the circuit to dilute the test substance instead of dispersing it. Given the extreme fluidic resistance (relative) of the neurite outgrowth channels, the level of dilution is insignificant. For experiments entailing the establishment of diffusion gradients, the aspiration method can be used to selectively treat one compartment, with flow cessation used to initiate the time-dependent evolution of the chemical gradient.
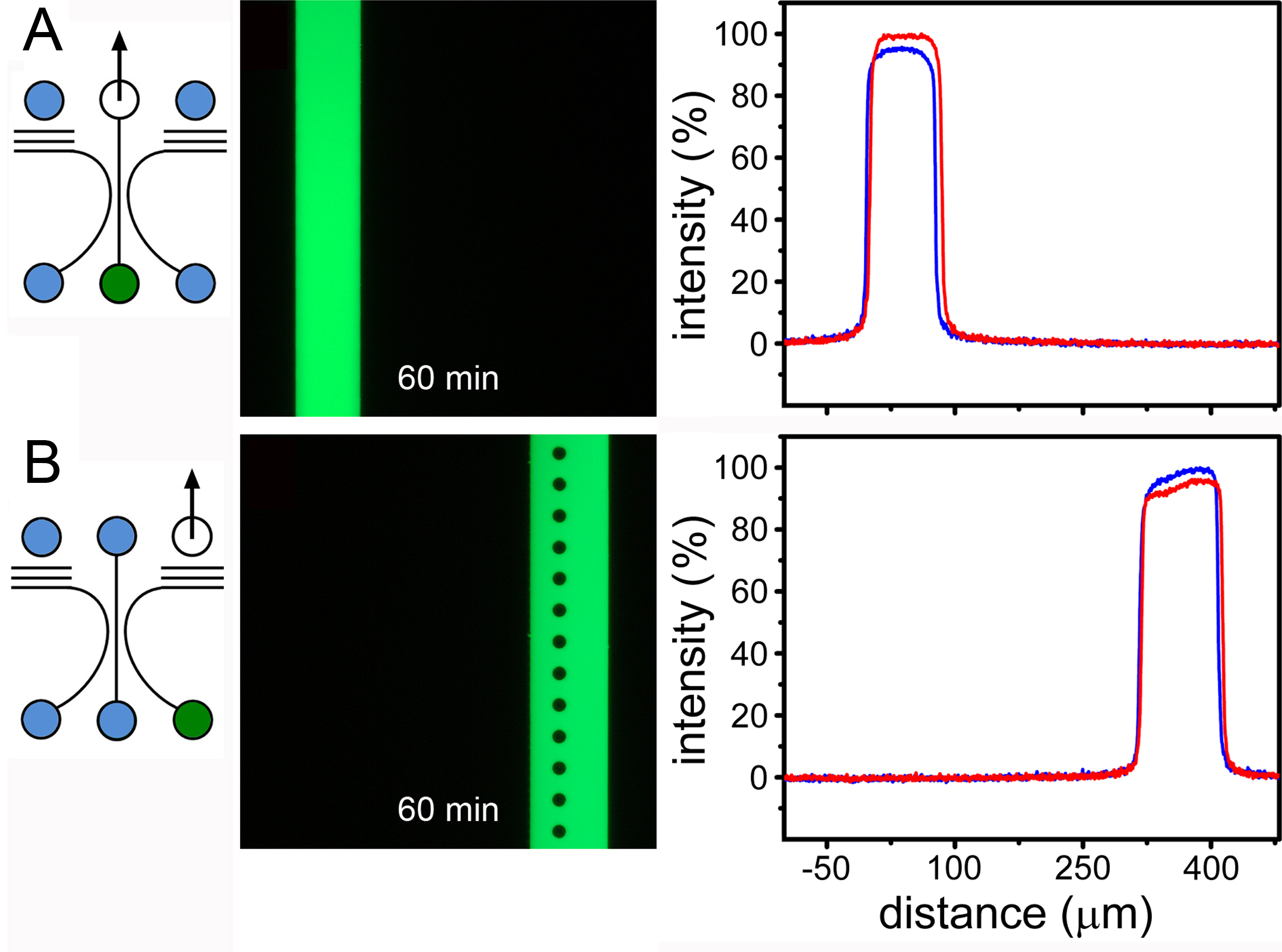
Figure 5. Selective fluidic treatments. A) Aspiration-driven selective treatments of the central and B) flanking compartments. The arrow indicates the single port used for aspiration. Treatments were established in 1 min (red lines) and were maintained for the duration of the experiment; 60 min (blue lines). Modified figure and legend reproduced with permission of the Royal Society of Chemistry (RSC)12. Click here to view larger image.
