Nervengewebe ist sehr komplex; eine heterogene Zellmischung, die räumlich in definierten Schichten und Fächer und mit Kunststoff-Konnektivität über Zell-Kontakte und vor allem über Axon und Dendriten Auswüchse bestellt wird. Neue Techniken sind erforderlich, um mehr experimentelle Freiheit, tiefere Einblicke und entwirren zentralen Mechanismen der Krankheit, Entwicklung und gesunde Funktion gewinnen verleihen. Die Campenot Kammer 1,2 und kürzlich mikroAusführungs 3,4 kann für die ex vivo Herstellung von vernetzten neuronalen Co-Kulturen mit der Fähigkeit, ihre Neuriten Auswüchse selektiv stören die verschiedenen somatischen Populationen und auch verwendet werden. Diese mikrofluidischen Bauteilen haben beispielsweise verwendet, um Axon-Degeneration und Regeneration zu studieren folgenden chemischen 5,6 oder Laser-Axotomie 6-8, Tauopathie 9, virale Verbreitung 10,11, und mRNA-Lokalisierung in Axone 4.
ent ">Um die Reichweite der Neurobiologen erstrecken, sind technologische Entwicklungen erforderlich, um minimalistische neuronalen Co-Kulturen vorzubereiten. Dies ermöglicht die Herauslösung des neuronalen Netzes für die Ermittlung des Systems mit einzelnen Zelle und subzellulären Präzision. Die Anforderung für eine minimale Zellzahlen eröffnet die Möglichkeit, seltenen Zelltypen, einschließlich dopaminergen Substantia nigra-Zellen relevant Parkinson, Spiralganglien aus dem Ohr, peripheren Neuronen zu analysieren, und Stammzellen. Darüber hinaus ist Mobilwirtschaft, die für die 3R-Initiative. Mit Hilfe dieser Mikrofluidik-Plattformen, große Bildschirme Toxizität oder andere Hochdurchsatz, können die Daten reichen Versuchsreihen, die Tier Neuronen betrachtet.
In diesem Papier Protokolle für die Herstellung und Verwendung einer Mikrofluid-Vorrichtung beschrieben. Mikrofluidik-Anordnungs in Kombination mit einem in situ Biomaterialienal Musterungsverfahren können für die Registrierung von hochvernetzten neuronalen Co-Kulturen mit minimalem Zellzahlen verwendet werden. Mikrofluidik-Anordnungs auf einem Differenzstrom-Ansatz 12-15, wobei mikro Traps in einem Fluidkreis (mit REM-Bildern in Fig. 1 dargestellt) auf der Basis positioniert. Der Pfad 0 → 1 die untere fluidische Widerstand (R 2> R 1) zum Transportieren Neuronen zu einer linearen Anordnung von mikrostrukturierten Öffnungen – die Einlässe zu den Neuriten-Auswuchs-Kanälen. Belegung der Falle durch eine einzelne Zelle lokal behindert den Durchfluss auf die Stromlinien zum Einfangen nachfolgenden Zellen in benachbarten Fallen abzulenken. Vollständige Belegung der Fallen in der Anordnung schaltet das fluidische Verhältnis (R 1> R 2), um die Stromlinien in den Serpentinenpfad (0 → 2) umzuleiten, um eine Bypass-Betriebsmodus für die Entfernung von überschüssigem Neuronen zu erzeugen.
<p class = "jove_content" fo: keep-together.within-page = "always">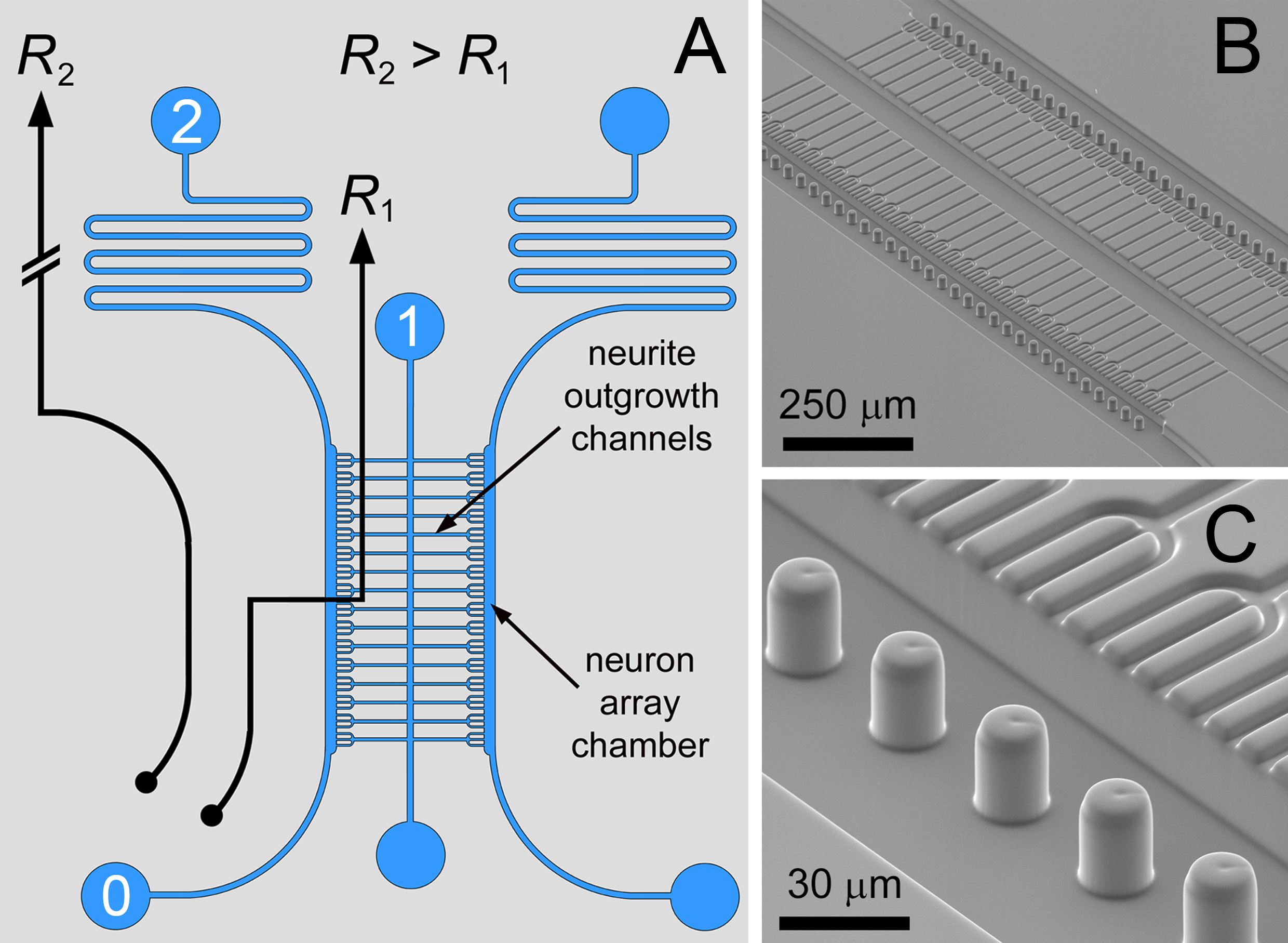
Abbildung 1. Mikrofluidik-Circuit. A) Die differentielle Widerstand Fluidkreis für einzelnes Neuron Anordnungs, mit flankierenden Kulturkammern von Neuritenwachstum Kanäle miteinander verbunden. B, C) SEM-Bilder der Doppelschicht compartmentalized Neuron Co-Kultur-Array mit Meniskus Pinning Mikrosäulen. Mit diesem Entwurf wurden Dreizack-förmigen Neuron Fangstrukturen verwendet, um Faszikulation der Neuriten Auswüchse zu fördern. Abbildung und Legende mit Genehmigung der Royal Society of Chemistry (RSC) wiedergegeben 12. Klicken Sie hier für eine größere Ansicht.
Die Herstellung von mikro neuronaler Netzwerke auf ebenen Substraten kann leicht erreicht werden (exampl es aus unserer Gruppe finden Frimat et al. 16 und Heike et al. 17). Allerdings Verkapselung bioaktiven Material Muster in PDMS-Geräten und mit der Anforderung des Mikrometer-Skala Ausrichtung der diese den mikrofluidischen Kanälen stellt eine große technische Herausforderung. In Abschnitt 3.1 wird ein Protokoll für die in dem Chip oder in situ Herstellung von Biomaterial-Muster dargestellt. Diese Muster ermöglichen Neuron Anmeldung bei längeren Zeitskalen und Kultur zu fördern Auswüchse zwischen Fächern. Meniskus-Pinning Mikrostrukturen verwendet werden, um eine so genannte Wasser-Maske mit dem Neuron Anordnen Sites und Neuritenwachstum Kanälen auszurichten. Das Wasser Maske schützt Adhäsionsmolekül Beschichtungen während der Plasmabehandlung, während exponierten Flächen werden aufgelöst, das Biomaterial Muster definieren. Zusätzlich sind Protokolle für die Zellkultur und für die fluidische Trennung für die selektive Behandlung der verschiedenen Co-Kultur Kammern notwendig ist.
ve_content "> Die Protokolle sind auf die benutzerfreundliche Prinzipien der Softlithographie für die Replikation von Poly (dimethylsiloxan) (PDMS) mikrofluidischen Vorrichtungen 18 zu nutzen. Auch in situ Biomaterial Musterung ist einfach, die Nutzung Verdunstung und Oberflächenspannung Phänomene und nur erfordern eine preiswerte Hand-Plasmaquelle. Mikrofluidik-Schaltung effektiv Programme Zelle Laderaum und spezielle Behandlungen machen diese Operationen nur eine Frage der Abgabe von Materialien in die richtige unteren Anschluss und Absaugen von oben. Auf diese Weise beabsichtigt ist, die Neurobiologen geben die Freiheit, in den eigenen Labors herzustellen und zu verwenden mikrofluidischen Bauteilen.