Tessuto neuronale è molto complesso; una miscela cellulare eterogenea che è spazialmente ordinato entro livelli e comparti definiti e con connettività di plastica tramite contatti cellulari e soprattutto tramite assoni e dendriti escrescenze. Nuove tecniche sono tenuti a conferire una maggiore libertà di sperimentazione per acquisire conoscenze più profonde e meccanismi a svelare centrali per malattia, lo sviluppo e la funzione sana. La camera Campenot 1,2 e più recentemente microfabricated realizzazioni 3,4 possono essere utilizzati per la preparazione di ex vivo neuronali co-colture in rete con la capacità di perturbare selettivamente le diverse popolazioni somatiche e anche loro escrescenze neuriti. Questi dispositivi microfluidici sono per esempio stati utilizzati per studiare assonale degenerazione e rigenerazione seguenti sostanze chimiche 5,6 o laser assotomia 6-8, tauopathy 9, la diffusione virale 10,11, e mRNA localizzazione in assoni 4.
ent ">Per estendere la portata dei neurobiologi, gli sviluppi tecnologici sono tenute a redigere neuronali co-culture minimalista. Ciò consente disentanglement della rete neuronale per la ricerca del sistema con singola cella e precisione sub-cellulare. Il requisito per il numero di cellule minime apre la possibilità di analizzare tipi di cellule rare, comprese dopaminergici della substantia nigra cellule pertinenti alla malattia di Parkinson, gangli spirale dall'orecchio, neuroni periferici, e cellule staminali. Al di là di questo, economia cellulare è rilevante per l'iniziativa 3R. Utilizzando queste piattaforme microfluidica, schermi di tossicità su larga scala o altro throughput elevato, i dati ricca serie sperimentali che richiedono neuroni di animali può essere considerato.
In questo documento sono descritti protocolli per la fabbricazione e l'uso di un dispositivo microfluidica. Arraying microfluidica in combinazione con un in situ biomateripatterning al metodo può essere utilizzato per la registrazione di co-colture neuronali altamente interconnesse utilizzando il numero di cellule minime. Arraying microfluidica è basato su un approccio differenziale di flusso 12-15, in cui trappole microstrutturata sono posizionati all'interno di un circuito fluidico (illustrato con immagini SEM di Figura 1). Il percorso 0 → 1 ha la resistenza fluida inferiore (R 2> R 1) per il trasporto di neuroni di una schiera lineare di aperture microstrutturati – le insenature ai canali crescita dei neuriti. Occupazione della trappola da una singola cellula impedisce localmente il flusso per deviare le linee di corrente per la cattura di celle successive in trappole vicini. Occupazione completa delle trappole nella matrice commuta il rapporto fluidico (R 1> R 2) per deviare le linee di flusso nel percorso a serpentina (0 → 2) per produrre un bypass di operazione per la rimozione dei neuroni in eccesso.
<p class = "jove_content" fo: keep-together.within-page = "always">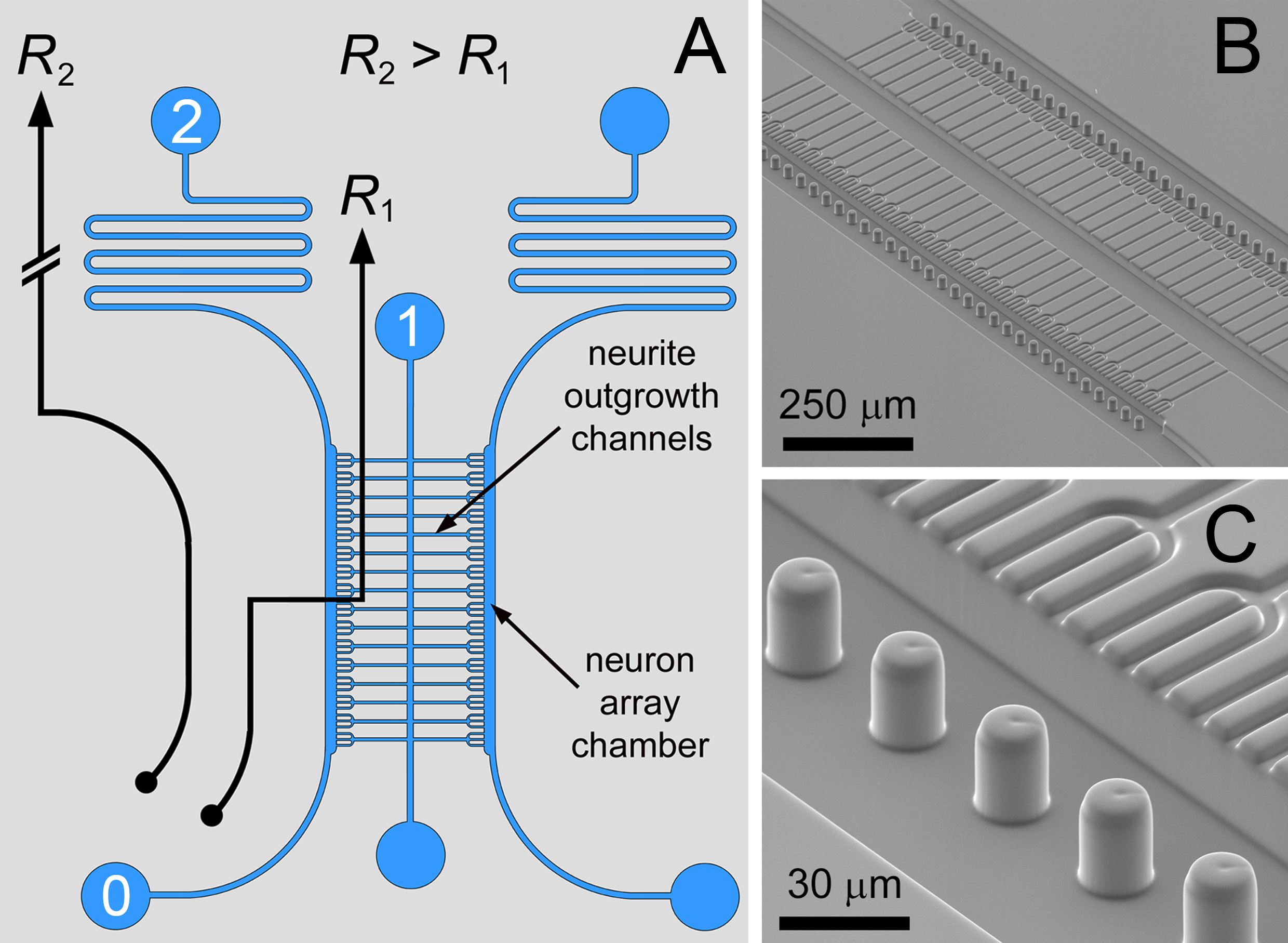
Figura 1. Microfluidic Circuit. A) Il circuito di resistenza differenziale di fluido per formare gli array singolo neurone, con accompagnamento camere di coltura collegati tra loro da canali crescita dei neuriti. B, immagini C) SEM del doppio strato neurone compartimenti matrice co-coltura con menisco pinning micropillars. Con questo progetto, le strutture neurone di cattura a forma di tridente sono stati utilizzati per promuovere la fascicolazione delle escrescenze neuriti. Figure e leggenda riprodotta con il permesso della Royal Society of Chemistry (RSC) 12. Clicca qui per vedere l'immagine ingrandita.
La preparazione delle reti neuronali micropatterned su substrati planari può facilmente essere ottenuto (per exampl es dal nostro gruppo, vedere Frimat et al. 16 e Heike et al. 17). Tuttavia, incapsulando materiale modelli bioattive all'interno dei dispositivi PDMS e con l'esigenza di allineamento micrometro scala di questi per i canali microfluidica rappresenta una grande sfida tecnica. Nella sezione 3.1 un protocollo per la a-chip, o in situ, preparazione di modelli biomateriale è presentato. Questi modelli consentono la registrazione dei neuroni durante tempi lunghi della cultura e promuovere escrescenze tra i compartimenti. Microstrutture menisco-pinning sono usati per allineare una cosiddetta maschera acqua con il neurone arraying siti e canali crescita dei neuriti. La maschera d'acqua protegge i rivestimenti molecole di adesione durante il trattamento al plasma, mentre le superfici esposte sono disintegrati per definire il modello di biomateriale. Inoltre, sono disponibili protocolli per colture cellulari e per l'isolamento fluidico necessaria per il trattamento selettivo dei vari comparti co-coltura.
ve_content "> I protocolli sono progettati per sfruttare i principi user-friendly di litografia soft per la replica di poli (dimetilsilossano) (PDMS) dispositivi microfluidici 18. Allo stesso modo, in situ biomateriale patterning è semplice, sfruttando evaporazione e superficiali fenomeni di tensione, e solo richiede una sorgente di plasma portatile poco costoso. Il circuito microfluidica efficace i programmi di carico e vano cella trattamenti specifici che queste operazioni semplicemente di erogazione di materiali nella porta inferiore corretta ed aspirando dall'alto. In questo modo, si intende dare le neurobiologists la libertà di preparare e utilizzare i dispositivi microfluidici nei propri laboratori.