神経組織は非常に複雑である。空間的に細胞接触を通じて、特に軸索と樹状突起増生によって定義層や区画内とプラスチックの接続と一緒に注文された異種の細胞の混合物である。新技術は、より深い洞察力や病気、開発、健康的な機能の中心UNRAVELメカニズムを獲得する大きな実験的な自由を与えることが必要である。 Campenot室1,2、最近微細加工の実施例3,4は、選択的に異なる体細胞集団と、それらの神経突起増生を撹乱する能力を備えたネットワーク接続された神経細胞の共培養のex vivo製造に使用することができる。これらのマイクロ流体デバイスは、たとえば、化学、5,6またはレーザー軸索切断6-8、タウオパチー9、ウイルス伝播10,11、および軸索4におけるmRNAの局在後の軸索の変性と再生を研究するために使用されてきた。
ENT」>神経生物学者の範囲を拡大するために、技術開発を最小限の神経細胞の共培養を準備する必要があります。これは、単一の細胞と細胞内の精度でシステムの調査のための神経回路網の解きほぐしを可能にします。最小限の細胞数の要件は、パーキンソン病、耳から螺旋神経節、末梢ニューロンに関連するドーパミン作動性黒質細胞を含む希少細胞タイプを分析し、幹細胞の可能性を開く。これ以外に、携帯経済は3Rのイニシアティブに関連しています。これらのマイクロ流体プラットフォームは、大規模な毒性スクリーニングまたは他のハイスループットを用いて、動物のニューロンを必要とするデータに富む一連の実験を考えることができる。
本論文では、マイクロ流体デバイスの製造および使用のためのプロトコルが記載されている。 その場 biomateri 内と組み合わせたマイクロ流体アレイ化他のパターニング方法は、最小限の細胞数を用いて高度に相互接続神経細胞の共培養の登録に使用することができる。マイクロ流体アレイ化は、差動フロー· アプローチの微細構造トラップは流体回路( 図1のSEM像と一緒に示されている)内に配置されることにより、12月15日 、に基づいています。神経突起伸長チャネルへの入口-パス0→1は、微細構造化アパーチャの直線状のアレイにニューロンを搬送するためのより低い流動抵抗(R 2> R 1)を有する。単一のセルによるトラップの占有率が局部的に近隣のトラップに、その後の細胞を捕捉するための流線をそらすために流れを妨げる。アレイ内のトラップの完全な占有は、過剰なニューロンの除去のための操作のバイパスモードを生成するために蛇行路(0→2)に流線をそらすために、流体の比(R 1> R 2)を切り替える。
<p claSS FO = "jove_content":キープtogether.withinページ= "常に">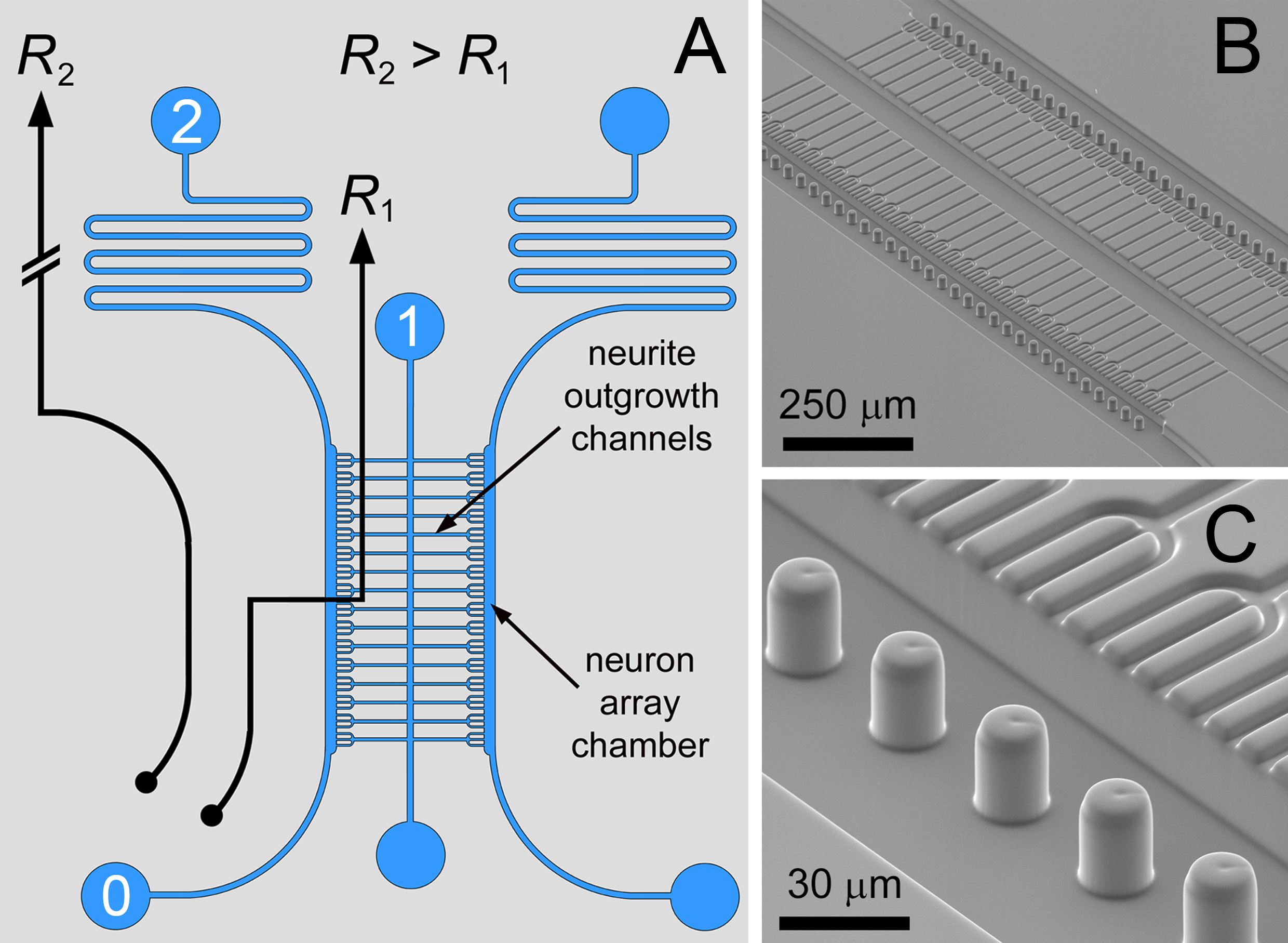
図1。マイクロ流体回路。マイクロピラーメニスカス固定有する二重層区画化ニューロン共培養アレイのA)神経突起伸長チャネルによって相互接続された培養チャンバに隣接有する単一ニューロン整列のために微分抵抗流体回路を、B、C)SEM画像である。この設計では、三叉状ニューロントラッピング構造は、神経突起増殖物の束形成を促進するために使用した。図や王立化学協会(RSC)の許可を得て複製伝説12。 拡大画像を表示するにはここをクリックしてください。
平面基板上のマイクロパターン神経回路網の製造は容易にexamplのために(達成することができ我々のグループからのESは、Frimat ら 16と平家ら 17)を参照してください。しかし、PDMSデバイス内およびマイクロ流体チャネルへのこれらのマイクロメートルスケール整列の要件を、生理活性物質パターンをカプセル化することは、主要な技術的課題を提起する。セクション3.1にするためのプロトコルは、チップ内、またはその場で 、生体材料のパターンの製造は、提示されている。これらのパターンは、長い培養時間スケールの間にニューロンの登録を有効にして、区画間の増生を促進する。メニスカスピニング微細構造ニューロン部位および神経突起伸長のチャンネルを配列して、所謂水マスクを整列させるために使用される。露出表面は生体材料のパターンを定義するために解砕されるのに対し、水マスクは、プラズマ処理の間、接着分子コーティングを保護する。また、プロトコルは、細胞培養のために、異なる共培養区画の選択的治療のために必要な流体の単離のために設けられている。
ve_content ">プロトコルは同様に、 その場での生体材料のパターニングに蒸発し、表面張力現象を利用し、簡単です。ポリレプリケーション(ジメチルシロキサン)(PDMS)マイクロ流体デバイス18のためのソフトリソグラフィーのユーザーフレンドリーな原則を活用するように設計されており、唯一の安価なハンドヘルド型プラズマ源を必要とする。マイクロ流体回路を効果的にプログラム正しい底部ポートに材料を分配し、上方から吸引するだけで問題これらの操作を行うセル負荷と区画特定の治療法。この方法では、神経生物学者を与えることを意図している自分の研究室でマイクロ流体デバイスを準備し、使用する自由。