

Summary
शाही सेना अनुक्रमण और जैव सूचना विज्ञान विश्लेषण माउस EMLcells की लिन-CD34 + और लिन-CD34- उप-जनसंख्या में काफी और विभिन्न व्यक्त प्रतिलेखन कारक की पहचान करने के लिए इस्तेमाल किया गया. ये प्रतिलेखन कारक स्वयं renewing लिन-CD34 + और आंशिक रूप से भेदभाव लिन-CD34- कोशिकाओं के बीच स्विच निर्धारित करने में महत्वपूर्ण भूमिका निभा सकता है.
Abstract
Hematopoietic स्टेम कोशिकाओं (HSCs) ऐसे ल्यूकेमिया और लिम्फोमा के रूप में कई रोगों में एक मरीज की hematopoietic प्रणाली के पुनर्निर्माण के लिए प्रत्यारोपण उपचार के लिए चिकित्सकीय उपयोग किया जाता है. HSCs आत्म नवीकरण और भेदभाव को नियंत्रित तंत्र elucidating अनुसंधान और नैदानिक उपयोगों के लिए HSCs के आवेदन के लिए महत्वपूर्ण है. हालांकि, यह कारण इन विट्रो में पैदा करना उनकी अक्षमता को HSCs की बड़ी मात्रा में प्राप्त करने के लिए संभव नहीं है. इस बाधा को दूर करने के लिए, हम इस अध्ययन के लिए एक मॉडल प्रणाली के रूप में, एक माउस अस्थि मज्जा व्युत्पन्न सेल लाइन, EML (erythroid, माइलॉयड, और Lymphocytic) सेल लाइन का इस्तेमाल किया.
शाही सेना अनुक्रमण (आरएनए Seq) तेजी से जीन अभिव्यक्ति के अध्ययन के लिए माइक्रोएरे को बदलने के लिए इस्तेमाल किया गया है. हम यहाँ EML सेल आत्म नवीकरण और भेदभाव के नियमन में संभावित महत्वपूर्ण कारकों की जांच के लिए शाही सेना Seq प्रौद्योगिकी के उपयोग की एक विस्तृत विधि की रिपोर्ट. इस पत्र में प्रदान प्रोटोकॉल को तीन भागों में बांटा गया है. पहला बराबरटी कैसे संस्कृति EML कोशिकाओं और अलग लिन-CD34 + और लिन-CD34- कोशिकाओं को बताते हैं. प्रोटोकॉल के दूसरे भाग में कुल शाही सेना की तैयारी और उच्च throughput अनुक्रमण के लिए बाद में पुस्तकालय निर्माण के लिए विस्तृत प्रक्रियाओं प्रदान करता है. पिछले भाग आरएनए Seq डेटा विश्लेषण के लिए विधि का वर्णन करता है और लिन-CD34 + और लिन-CD34- कोशिकाओं के बीच विभिन्न व्यक्त प्रतिलेखन कारक की पहचान करने के लिए डेटा का उपयोग करने के लिए बताते हैं. सबसे महत्वपूर्ण विभिन्न व्यक्त प्रतिलेखन कारक EML सेल आत्म नवीकरण और भेदभाव को नियंत्रित संभावित कुंजी नियामकों होने की पहचान की गई. इस पत्र की चर्चा खंड में, हम इस प्रयोग के सफल प्रदर्शन के लिए महत्वपूर्ण कदम पर प्रकाश डाला.
संक्षेप में, इस पत्र EML कोशिकाओं में आत्म नवीकरण और भेदभाव के संभावित नियामकों की पहचान करने के लिए शाही सेना Seq प्रौद्योगिकी का उपयोग करने का एक तरीका प्रदान करता है. पहचान महत्वपूर्ण कारक इन विट्रो और मैं में नीचे की ओर कार्यात्मक विश्लेषण के अधीन हैंएन विवो.
Introduction
Hematopoietic स्टेम सेल वयस्क अस्थि मज्जा आला में मुख्य रूप से रहते हैं कि दुर्लभ रक्त कोशिकाओं रहे हैं. वे रक्त की भरपाई करने के लिए आवश्यक कोशिकाओं और प्रतिरक्षा प्रणाली 1 के उत्पादन के लिए जिम्मेदार हैं. स्टेम कोशिकाओं का एक प्रकार के रूप में, HSCs आत्म नवीकरण और भेदभाव दोनों में सक्षम हैं. HSCs के भाग्य का फैसला है कि नियंत्रण तंत्र elucidating, आत्म नवीकरण या भेदभाव या तो ओर, रक्त रोग शोध और नैदानिक उपयोग 2 के लिए HSCs के हेरफेर पर बहुमूल्य मार्गदर्शन प्रदान करेगी. शोधकर्ताओं द्वारा सामना की एक समस्या यह HSCs बनाए रखा और एक बहुत ही सीमित हद तक इन विट्रो में विस्तार किया जा सकता है; उनकी संतान के विशाल बहुमत आंशिक रूप से संस्कृति 2 में भेदभाव कर रहे हैं.
एक जीनोम व्यापक पैमाने पर आत्म नवीकरण और भेदभाव की प्रक्रियाओं को नियंत्रित कि प्रमुख नियामकों की पहचान करने के लिए, हम एक मॉडल प्रणाली के रूप में एक माउस आदिम hematopoietic पूर्वज सेल लाइन EML इस्तेमाल किया. गुसेल लाइन murine अस्थि मज्जा 3,4 से प्राप्त किया गया है. विभिन्न विकास कारकों से तंग आ चुके हैं, तो EML कोशिकाओं इन विट्रो 5 में एर्य्थ्रोइद, माइलॉयड, और ल्य्म्फोइड कोशिकाओं में अंतर कर सकते हैं. महत्वपूर्ण बात है, इस सेल लाइन संस्कृति के माध्यम से उनके multipotentiality बनाए रखना अभी भी स्टेम सेल कारक (एस सी एफ) युक्त और में बड़ी मात्रा में प्रचारित किया जा सकता है. EML कोशिकाओं स्वयं renewing लिन-एससीए + CD34 + के उप-जनसंख्या में विभाजित है और आंशिक रूप से सतह मार्कर CD34 और एससीए 6 पर आधारित लिन-एससीए-CD34- कोशिकाओं विभेदित किया जा सकता है. अल्पकालिक HSCs, एससीए + CD34 + कोशिकाओं के लिए इसी प्रकार आत्म नवीकरण के लिए सक्षम हैं. तेजी लिन-एससीए + CD34 + और लिन-एससीए-CD34- कोशिकाओं की एक मिश्रित आबादी पुनर्जन्म और 6 पैदा करना जारी रख सकते हैं एस सी एफ, लिन-एससीए + CD34 + कोशिकाओं के साथ व्यवहार करते हैं. दो आबादी आकृति विज्ञान में समान हैं और सी-किट mRNA और प्रोटीन 6 के समान स्तर है. लिन-एससीए-CD34- कोशिकाओं आईएल -3 के बजाय एस सी एफ 3 युक्त मीडिया में प्रचार करने में सक्षम हैं. Unveilinजी hematopoiesis दौरान जल्दी विकास संक्रमण में सेलुलर और आणविक तंत्र की बेहतर समझ प्रदान करेगी EML सेल भाग्य निर्णय में महत्वपूर्ण नियामकों.
आत्म नवीकरण लिन-एससीए + CD34 + और आंशिक रूप से भेदभाव लिन-एससीए-CD34- कोशिकाओं के बीच अंतर्निहित आणविक मतभेद की जांच करने के लिए, हम विभिन्न व्यक्त जीनों की पहचान करने के लिए शाही सेना Seq इस्तेमाल किया. प्रतिलेखन कारक सेल भाग्य का निर्धारण करने में महत्वपूर्ण हैं के रूप में विशेष रूप से, हम, प्रतिलेखन कारक पर ध्यान केंद्रित. आरएनए Seq प्रोफ़ाइल और जीनोम 7,8 से लिखित RNAs यों (NGS) प्रौद्योगिकियों अगली पीढ़ी के अनुक्रमण की क्षमताओं का उपयोग एक हाल ही में विकसित दृष्टिकोण है. संक्षेप में, कुल शाही सेना है पाली-एक प्रारंभिक template.The शाही सेना टेम्पलेट फिर रिवर्स ट्रांसक्रिपटेस का उपयोग सीडीएनए में बदल जाता है के रूप में चुना और खंडित. सीडीएनए पुस्तकालय के निर्माण के लिए बरकरार, गैर अपमानित शाही सेना का उपयोग, पूर्ण लंबाई शाही सेना टेप नक्शा करने के क्रम में महत्वपूर्ण है. पुर के लिएअनुक्रमण का ढोंग, विशिष्ट एडाप्टर दृश्यों सीडीएनए के दोनों सिरों को जोड़ रहे हैं. फिर, ज्यादातर मामलों में, सीडीएनए अणुओं पीसीआर द्वारा परिलक्षित कर रहे हैं और एक उच्च throughput ढंग से अनुक्रम.
अनुक्रमण के बाद, जिसके परिणामस्वरूप संदर्भ जीनोम और एक transcriptome डेटाबेस के लिए गठबंधन किया जा सकता है पढ़ता है. की संख्या संदर्भ जीन को मानचित्र में गिना जाता है और इस जानकारी जीन अभिव्यक्ति के स्तर का अनुमान किया जा सकता है कि पढ़ता है. यह भी गैर-मॉडल जीवों 9 में transcriptomes के अध्ययन को सक्षम करने, एक संदर्भ जीनोम के बिना नए सिरे से इकट्ठा किया जा सकता पढ़ता है. आरएनए-सेक तकनीक भी ब्याह isoforms 10-12, उपन्यास टेप 13 और जीन fusions 14 का पता लगाने के लिए इस्तेमाल किया गया है. प्रोटीन कोडिंग जीन का पता लगाने के अलावा, आरएनए Seq भी ऐसे ही गैर-कोडिंग RNAs के प्रतिलेखन स्तर उपन्यास का पता लगाने और विश्लेषण करने के लिए इस्तेमाल किया जा सकता है लंबे समय siRNA आदि 18 आरएनए 15,16, microRNA 17, गैर-कोडिंग. क्योंकि टी कीइस विधि से वह शुद्धता, यह एकल nucleotide विविधताओं 19,20 का पता लगाने के लिए उपयोग किया गया है.
आरएनए Seq प्रौद्योगिकी के आगमन से पहले, माइक्रोएरे जीन अभिव्यक्ति प्रोफाइल विश्लेषण करने के लिए इस्तेमाल किया मुख्य विधि था. पूर्व डिजाइन जांच संश्लेषित और बाद में एक माइक्रोएरे स्लाइड 21 फार्म करने के लिए एक ठोस सतह से जुड़े होते हैं. mRNA के निकाले और सीडीएनए में बदल जाती है. रिवर्स प्रतिलेखन प्रक्रिया के दौरान, fluorescently लेबल न्यूक्लियोटाइड सीडीएनए में शामिल कर रहे हैं और सीडीएनए माइक्रोएरे स्लाइड पर संकरित किया जा सकता है. एक विशिष्ट स्थान से एकत्र संकेत की तीव्रता उस स्थान 21 पर विशिष्ट जांच के लिए बाध्य सीडीएनए की राशि पर निर्भर करता है. आरएनए Seq तकनीक के साथ तुलना में, माइक्रोएरे कई सीमाएं हैं. आरएनए Seq प्रौद्योगिकी इसके उपयोग करते समय जो सीमा सापेक्ष उच्च पृष्ठभूमि स्तर पर उपन्यास टेप पता लगाने में सक्षम है, जबकि पहले, माइक्रोएरे, जीन एनोटेशन के पूर्व मौजूदा ज्ञान पर निर्भर करता है जीईपूर्वोत्तर अभिव्यक्ति स्तर कम है. कारण पृष्ठभूमि और संकेतों की संतृप्ति के लिए, माइक्रोएरे की सटीकता दोनों अत्यधिक और नीच व्यक्त जीनों 7,22 के लिए सीमित है, जबकि इसके अलावा, आरएनए Seq प्रौद्योगिकी, पहचान (8,000 गुना) 7 की बहुत अधिक गतिशील रेंज है. अंत में, माइक्रोएरे जांच एक नमूना 23 के भीतर अलग टेप के रिश्तेदार अभिव्यक्ति के स्तर की तुलना जब परिणाम कम विश्वसनीय बनाने के जो उनके संकरण क्षमता में भिन्न होते हैं. आरएनए Seq माइक्रोएरे पर कई फायदे हैं, अपने डेटा विश्लेषण जटिल है. यह कई शोधकर्ताओं ने अभी आरएनए seq के बजाय माइक्रोएरे का उपयोग करने वाले कारणों में से एक है. विभिन्न जैव सूचना विज्ञान उपकरण आरएनए Seq डाटा प्रोसेसिंग और विश्लेषण 24 के लिए आवश्यकता होती है.
कई अगली पीढ़ी के अनुक्रमण (NGS) प्लेटफॉर्म के अलावा, 454, Illumina, ठोस और आयन टोरेंट सबसे व्यापक रूप से इस्तेमाल कर रहे हैं. 454 पहला वाणिज्यिक NGS मंच था. अन्य अनुक्रमण प्लेटफार्मों के विपरीतऐसे Illumina और ठोस रूप में, 454 मंच पढ़ पाएंगे उत्पन्न लंबाई 25 (औसत 700 आधार पढ़ता है). कारण अब उच्च क्षमता 25 इकट्ठा उनके लिए transcriptiome के प्रारंभिक लक्षण वर्णन के लिए बेहतर हैं पढ़ता है. 454 मंच के मुख्य नुकसान अनुक्रम का megabase प्रति अपनी उच्च लागत है. उत्पन्न Illumina और ठोस प्लेटफार्मों बढ़ी संख्या और कम लंबाई के साथ पढ़ता है. अनुक्रम की megabase प्रति लागत 454 मंच की तुलना में काफी कम है. कारण Illumina और ठोस प्लेटफार्मों के लिए पढ़ता कम की बड़ी संख्या के लिए, डेटा विश्लेषण और अधिक computationally गहन है. आयन टोरेंट मंच के लिए अनुक्रमण के लिए साधन और अभिकर्मकों की कीमत सस्ती है और अनुक्रमण समय 25 कम है. हालांकि, त्रुटि दर और अनुक्रम की megabase प्रति लागत अधिक Illumina और ठोस प्लेटफार्मों की तुलना कर रहे हैं. विभिन्न प्लेटफार्मों के अपने फायदे और नुकसान हैं और डेटा विश्लेषण के लिए विभिन्न तरीकों की आवश्यकता होती है. पीएलएtform अनुक्रमण उद्देश्य और धन की उपलब्धता के आधार पर चुना जाना चाहिए.
इस पत्र में, हम एक उदाहरण के रूप Illumina आरएनए Seq मंच ले. हम EML सेल आत्म नवीकरण और भेदभाव में प्रमुख नियामकों की जांच के लिए एक मॉडल प्रणाली के रूप में EML सेल का इस्तेमाल किया, और अभिव्यक्ति के स्तर गणना और उपन्यास प्रतिलिपि का पता लगाने के लिए शाही सेना Seq पुस्तकालय निर्माण और डेटा विश्लेषण का एक विस्तृत तरीकों प्रदान की. हम EML मॉडल प्रणाली 2 में आरएनए seq के अध्ययन, जब कार्यात्मक परीक्षण (जैसे shRNA पछाड़ना) hematopoietic भेदभाव की प्रारंभिक अवस्था के आणविक तंत्र को समझने में एक शक्तिशाली तरीका प्रदान के साथ मिलकर कि हमारे पिछले प्रकाशन में दिखाया गया है, और एक के रूप में सेवा कर सकते हैं सामान्य में सेल आत्म नवीकरण और भेदभाव के विश्लेषण के लिए मॉडल.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. EML सेल संस्कृति और सिस्टम और छंटनी चुंबकीय सेल का प्रयोग लिन-CD34 + और लिन-CD34- प्रकोष्ठों के पृथक्करण प्रतिदीप्ति सक्रिय सेल छंटनी विधि
- स्टेम सेल कारक संग्रह के लिए बच्चे हम्सटर गुर्दे (BHK) सेल संस्कृति माध्यम की तैयारी:
- 37 डिग्री सेल्सियस एक सेल कल्चर इनक्यूबेटर में, 5% सीओ 2 में 25 सेमी 2 कुप्पी (1 टेबल) में 10% FBS युक्त DMEM माध्यम में संस्कृति BHK कोशिकाओं.
- कोशिकाओं 80 बड़े होते हैं - 90% संगम, पीबीएस के 10 एमएल के साथ एक बार कोशिकाओं धो लो. Monolayer को 0.25% trypsin-EDTA समाधान के 5 मिलीलीटर जोड़ें और कोशिकाओं को अलग कर रहे हैं जब तक कमरे के तापमान (आरटी) में 1-5 मिनट के लिए कोशिकाओं को सेते हैं.
- Pipet नीचे धीरे समाधान और कोशिकाओं के clumps को तोड़ने के लिए. Trypsin गतिविधि को रोकने के लिए कुप्पी को पूरा DMEM के 5 मिलीलीटर जोड़ें. आरटी पर 5 मिनट के लिए 200 XG पर centrifugation द्वारा कोशिकाओं को इकट्ठा.
- मध्यम निकालें और ताजा BHK सेल संस्कृति के माध्यम से 10 एमएल में सेल गोली resuspend. स्थानांतरण 2 एक नया 75 सेमी 2 फ्लास्क कदम 1.1.4 से सेल निलंबन के मिलीलीटर और कुप्पी को ताजा BHK सेल संस्कृति के माध्यम से 48 मिलीलीटर जोड़ें.
- संस्कृति बेडरूम दो दिनों के लिए कोशिकाओं और संस्कृति के माध्यम से इकट्ठा. पारित होने के एक 0.45 माइक्रोन फिल्टर के माध्यम से मध्यम. आगे उपयोग करें जब तक -20 डिग्री सेल्सियस में मध्यम स्टोर.
- EML सेल संस्कृति:
- एक सेल कल्चर इनक्यूबेटर में 37 डिग्री सेल्सियस, 5% सीओ 2 BHK सेल संस्कृति माध्यम (1 टेबल) युक्त EML बुनियादी माध्यम में संस्कृति EML कोशिकाओं (निलंबन में).
- शिखर घनत्व कम से कम 6 x 10 5 कोशिकाओं / एमएल के साथ कम सेल घनत्व (0.5-5 x 10 5 कोशिकाओं / एमएल) में EML कोशिकाओं को बनाए रखने. 1 के अनुपात में हर 2-3 दिनों कोशिकाओं को विभाजित: 5. पारित होने EML कोशिकाओं धीरे और 10 पीढ़ियों के लिए passaging के बाद संस्कृति त्यागें.
- वंश सकारात्मक कोशिकाओं की कमी:
- 200 XG लिए पर centrifugation द्वारा EML कोशिकाओं फसलआर 5 मिनट और पीबीएस के साथ एक बार कोशिकाओं धो लो. 5 मिनट के लिए 200 XG पर centrifugation द्वारा कोशिकाओं को इकट्ठा.
- पीबीएस के साथ कोशिकाओं Resuspend और एक hemocytometer साथ कोशिकाओं गिनती. (सेल अलगाव प्रणाली के प्रदाता द्वारा की पेशकश निर्देश देखें) कोशिकाओं की संख्या के अनुसार बाद में सेल जुदाई चरण में एंटीबॉडी एकाग्रता का निर्धारण.
- वंश नकारात्मक (Lin-) पृथक वंश एंटीबॉडी कॉकटेल (बायोटिन संयुग्मित मोनोक्लोनल एंटीबॉडी के कॉकटेल का उपयोग कोशिकाओं CD5, CD45R (B220), CD11b, विरोधी जीआर -1 (ly-6G / सी), 7-4 और टेर-119 ) और निर्माता के निर्देशों के अनुसार एक चुंबकीय सक्रिय सेल छँटाई प्रणाली.
- लिन-CD34 + और लिन-CD34- कोशिकाओं के पृथक्करण:
- 5 मिनट के लिए 200 XG पर कदम 1.3.3 से Lin- कोशिकाओं नीचे स्पिन. पीबीएस के साथ सेल गोली resuspend और एक hemocytometer साथ कोशिकाओं गिनती.
- FACS बफर के साथ कोशिकाओं को दो बार धोएं और 200 XG पर गोली कोशिकाओं5 मिनट के लिए.
- क्रमशः संख्या 1, 2, 3, 4, 5 के साथ पाँच 1.5 मिलीलीटर microcentrifuge ट्यूबों लेबल. 10 6 कोशिकाओं (ट्यूब प्रति 10 6 कोशिकाओं) प्रति 100 μl FACS बफर के साथ कोशिकाओं Resuspend.
- ट्यूब 1 और ट्यूब से 2 विरोधी माउस CD34 FITC एंटीबॉडी का 1 माइक्रोग्राम प्रति जोड़ें और धीरे ट्यूब मिश्रण.
- अंधेरे में 1 घंटे के लिए 4 डिग्री सेल्सियस पर सभी ट्यूबों को सेते हैं.
- ट्यूब 1, एपीसी संयुग्मित वंश कॉकटेल एंटीबॉडी के 0.25 ट्यूब से 3 पीई संयुग्मित विरोधी Sca1 एंटीबॉडी की माइक्रोग्राम प्रति, और 20 μl करने के लिए वंश कॉकटेल एंटीबॉडी एपीसी संयुग्मित के पीई संयुग्मित विरोधी Sca1 एंटीबॉडी का 0.25 माइक्रोग्राम प्रति और 20 μl जोड़ें ट्यूब 4.
- धीरे सभी ट्यूबों मिक्स और अंधेरे में एक अतिरिक्त 30 मिनट के लिए 4 डिग्री सेल्सियस पर कोशिकाओं सेते हैं.
- कोशिकाओं के लिए FACS बफर के 300 μl जोड़ें और 5 मिनट के लिए 200 XG पर कोशिकाओं नीचे स्पिन.
- तीन बार के लिए FACS बफर के 500 μl के साथ कोशिकाओं को धो लें.
- FACS के नौकरशाह के 500 μl में सेल गोली Resuspendffer.
- मुआवजे की स्थापना के लिए ट्यूब 2, 3, 4, और 5 में कोशिकाओं का प्रयोग करें. FACS Aria का उपयोग ट्यूब 1 में लिन-एससीए + CD34 + और लिन-एससीए-CD34- कोशिकाओं को अलग.
2. उच्च throughput अनुक्रमण के लिए शाही सेना की तैयारी और पुस्तकालय निर्माण
- अलगाव, गुणवत्ता विश्लेषण और शाही सेना की मात्रा का ठहराव:
- विनिर्माण 'प्रोटोकॉल के बाद लिन-CD34 + और क्रमशः TRIzol का उपयोग लिन-CD34- कोशिकाओं से कुल शाही सेना निकालें.
- निर्माण के प्रोटोकॉल के बाद दूषित डीएनए का उपयोग deoxyribonuclease मैं (DNase मैं) निकालें. वैकल्पिक रूप से, आगे उपयोग के लिए इस चरण में -80 डिग्री सेल्सियस पर शाही सेना की दुकान.
- आपूर्तिकर्ता द्वारा की पेशकश निर्देश के अनुसार Bioanalyzer उपयोग कर कुल शाही सेना की गुणवत्ता का आकलन. शाही सेना वफ़ादारी संख्या (Rin) 9 से बीर के साथ शाही सेना के नमूने का प्रयोग करें.
- पुस्तकालय निर्माण और उच्च throughput अनुक्रमण:
नोट: इस प्रोटोकॉल Illumina मंच का उपयोग आरएनए Seq वर्णन करता है. के लिएअन्य अनुक्रमण प्लेटफार्मों, अलग पुस्तकालय तैयारी के तरीकों के लिए आवश्यक हैं.- पुस्तकालय तैयारी के लिए नमूना प्रति उच्च गुणवत्ता कुल शाही सेना के 0.1-4 माइक्रोग्राम प्रति का उपयोग करें. आम तौर पर कुल शाही सेना के 2 माइक्रोग्राम प्रति 10 5 EML कोशिकाओं से निकाला जा सकता है.
- शाही सेना शुद्धि और विखंडन, पहला और दूसरा किनारा सीडीएनए संश्लेषण, अंत मरम्मत, 3 के लिए एक शाही सेना अनुक्रमण नमूना तैयार प्रणाली का प्रयोग करें 'प्रदाता के निर्देशों से विस्तृत मानक प्रक्रियाओं का पालन, adenylation, अनुकूलक बंधाव और पीसीआर प्रवर्धन समाप्त होता है.
- सकारात्मक oligo-डीटी चुंबकीय मोती का उपयोग Pólya mRNA के चयन और mRNA टुकड़ा.
- सीडीएनए प्राप्त करने और बाद में डबल असहाय सीडीएनए उत्पन्न करने के लिए सीडीएनए का दूसरा किनारा संश्लेषण करने यादृच्छिक प्राइमरों का उपयोग रिवर्स प्रतिलेखन प्रदर्शन करना.
- 3 'overhangs और 5 भरना' निकालें डीएनए पोलीमरेज़ द्वारा overhangs. Adenylate 3 'एक दूसरे से ligating से सीडीएनए टुकड़े को रोकने के लिए समाप्त हो जाती है.
- DscDNA के दोनों सिरों को मल्टीप्लेक्स अनुक्रमण एडेप्टर जोड़ें. डीएनए टुकड़े के संवर्धन के लिए पीसीआर प्रदर्शन करना.
- एक स्पेक्ट्रोफोटोमीटर का उपयोग पुस्तकालय की एकाग्रता के बारे में जानकारी प्राप्त करने के लिए A260 / A280 उपाय.
- पुस्तकालय गुणवत्ता का आकलन और एक Bioanalyzer का उपयोग डीएनए टुकड़े का आकार सीमा को मापने.
3. डेटा विश्लेषण
इस हिस्से में इस्तेमाल किया सॉफ्टवेयर के संदर्भ के लिए, (तालिका 2) देखें.
- बहाव के विश्लेषण के लिए डेटा फ़ाइल प्रसंस्करण:
- .bcl (बेस कॉल फ़ाइल) कन्वर्ट casava सॉफ्टवेयर (Illumina, संस्करण 1.8.2) का उपयोग कर फ़ाइल .fastq फ़ाइल.
- लिनक्स सिस्टम में 'टर्मिनल' आग. एक Illumina HiSeq2000 अनुक्रमण मशीन से डेटा फ़ाइल में डेटा फ़ोल्डर में जाओ. परिणाम फ़ोल्डर 'NASboy1 / JiaqianLabData / HiSeq_RUN / 2013_07_11 / 130627_SN860_0309_A_2013-166_H0PW9ADXX /', प्रकार है मान लीजिएचित्रा S1A में आदेश, और में डेटा फ़ोल्डर दर्ज करें.
- लिनक्स सिस्टम में casava 1.8.2 स्थापित करें. , Outputfolder 'unaligned' है मान लीजिए परिवर्तित करने के लिए विन्यास फाइल तैयार करने के लिए चित्रा S1B में आदेश का उपयोग करें. केवल एक .fastq फ़ाइल प्रत्येक नमूने के लिए बनाया जाता है यह सुनिश्चित करने के लिए --fastq क्लस्टर गिनती 0 विकल्प का प्रयोग करें. उत्पन्न .fastq फ़ाइल .gz प्रारूप में है. बहाव के विश्लेषण (चित्रा S1B) के लिए इसे खोल देना.
- 'Unaligned' फ़ोल्डर उत्पन्न किया गया है के बाद, 'unaligned' फ़ोल्डर (चित्रा S1C) के पास जाओ.
- बदलने की प्रक्रिया शुरू करने के लिए चित्रा S1D में आदेश का उपयोग करें. '-j' पैरामीटर का उपयोग किया जाएगा कि सीपीयू संख्या सप्लाई करती है.
- प्रणाली परिवर्तित करने की प्रक्रिया समाप्त होने के बाद, 'unaligned' फ़ोल्डर (चित्रा S1E) के तहत परिणाम फ़ोल्डर में जाओ.
- <चित्रा S1F में आदेश का उपयोग करें/ Strong> के प्रत्येक नमूने फ़ोल्डर के अंतर्गत .fastq फ़ाइल में .fastq.gz फ़ाइल दबाव हटाना.
- .bcl (बेस कॉल फ़ाइल) कन्वर्ट casava सॉफ्टवेयर (Illumina, संस्करण 1.8.2) का उपयोग कर फ़ाइल .fastq फ़ाइल.
- उपन्यास टेप का पता लगाने और tuxedo सुइट 26 का उपयोग कर अभिव्यक्ति के स्तर का मूल्यांकन:
- आरएनए Seq (से प्राप्त UCSC संस्करण mm9, माउस संदर्भ जीनोम को पढ़ता बनती अंत मानचित्र http://cufflinks.cbcb.umd.edu/igenomes.html का उपयोग करता है) Tophat सॉफ्टवेयर का उपयोग (संस्करण 1.3.3) 27, Bowtie मैपर (संस्करण 0.12.7) 28 पढ़ा. Tophat अभिव्यक्ति के स्तर का आकलन सटीकता में सुधार करने के लिए "-नहीं-उपन्यास-juncs" विकल्प के साथ आपूर्ति की है.
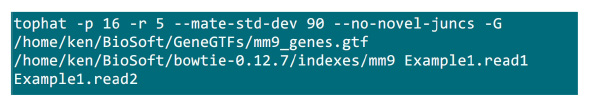
- मानचित्रण प्रक्रिया लागू किया जाएगा जहां एक फ़ोल्डर में .fastq फ़ाइलों रखो. एक बनती अंत अनुक्रमण नमूना के लिए 2 .fastq फ़ाइलें (Example1.read1, Example1.read2 को नाम बदलने) कर रहे हैं मान लीजिए मानचित्रण (सिस्टम सेटिंग के अनुसार मानकों समायोजित) करना चित्रा S2 में आदेश का उपयोग करें."-p" पैरामीटर का उपयोग किया जाएगा कि सीपीयू संख्या सप्लाई करती है. "आर" और "-mate एसटीडी देव" मापदंडों पुस्तकालय क्यूसी से प्राप्त या (चित्रा S2) गठबंधन का एक सबसेट से पढ़ता निष्कर्ष निकाला जा सकता है.
- कफ़लिंक सॉफ्टवेयर का उपयोग कर शाही सेना टेप में पढ़ता मैप किया (संस्करण 1.3.0) 29 इकट्ठे. एनोटेशन ज्ञात जीन की फ़ाइल (Tophat द्वारा इस्तेमाल किया ही .gtf फ़ाइल) और Tophat द्वारा उत्पादित .bam फ़ाइल का उपयोग कर चला कफ़लिंक.
- Tophat चलना समाप्त होने के बाद, एक ही फ़ोल्डर में, transcriptome और अनुमान प्रतिलिपि अभिव्यक्ति के स्तर के निर्माण के लिए कफ़लिंक चलाने के लिए चित्रा S3A में आदेश का उपयोग करें. 'GenomeSeqMM9' फ़ोल्डर में 'mm9_repeatMasker.gtf' और जीनोम अनुक्रम फ़ाइलों UCSC जीनोम ब्राउज़र से प्राप्त किया जा सकता है.
- परिणामस्वरूप genes.expr और transcripts.expr फ़ाइलों जीन और टेप (isoforms) की अभिव्यक्ति मूल्य होते हैं. कॉपी और पेस्टएक एक्सेल में फाइल सामग्री फाइल और स्प्रेडशीट आवेदन (चित्रा S3B) के साथ हेरफेर.
- उपन्यास टेप की पहचान करने के क्रम में संदर्भ 'mm9_genes.gtf' फ़ाइल के परिणामस्वरूप 'transcripts.gtf' फ़ाइल तुलना करने के लिए चित्रा S3C में आदेश का उपयोग करें.
- परिणामस्वरूप .tmap फ़ाइल तुलना परिणाम होता है. कॉपी और एक एक्सेल फाइल करने के लिए फ़ाइल की सामग्री चस्पा और स्प्रेडशीट आवेदन के साथ हेरफेर. कक्षा कोड के साथ देखिए 'यू' (चित्रा s3d) प्रदान की फ़ाइल .gtf संदर्भ की तुलना में 'उपन्यास' के रूप में माना जा सकता है.
नोट: मूल्यों 0.1 के तहत कर रहे हैं अगर बहाव विश्लेषण सुविधा के लिए, 0.1 के लिए FPKM मूल्यों की स्थापना की.
नोट: चरण 3.2.3 - 3.2.6 उपन्यास टेप 'अभिव्यक्ति आकलन की सटीकता में सुधार करना चाहते हैं के लिए वैकल्पिक है. मानचित्रण और transcriptome निर्माण अनुसंधान करने की आवश्यकता है, क्योंकि यह एक बहुत लंबे समय में समय लगेगासंयुक्त राष्ट्र के एक बार से अधिक.
- डिफ़ॉल्ट मापदंडों का उपयोग Tophat चलाने और फिर चित्रा S3E में आदेश का उपयोग कर उत्पन्न .gtf फ़ाइल को कफ़लिंक चलाते हैं.
- चित्रा S3F में आदेश का उपयोग संदर्भ जीनोम .gtf फ़ाइल के परिणामस्वरूप .gtf फ़ाइल की तुलना करें.
- कदम 3.2.2.4 में वर्णित के रूप में हुई .tmap फ़ाइल को पार्स. कॉपी और एक एक्सेल फाइल करने के लिए फ़ाइल की सामग्री चस्पा और स्प्रेडशीट आवेदन के साथ हेरफेर. कक्षा कोड के साथ देखिए 'यू' के रूप में माना जा सकता है 'उपन्यास' प्रदान की फ़ाइल .gtf संदर्भ की तुलना में.
- कदम 3.2.5 के बाद, संदर्भ .gtf फ़ाइल के रूप में इस्तेमाल किया जा सकता है जो फ़ोल्डर में एक .combined.gtf फ़ाइल नहीं है. उपन्यास टेप की एक अधिक सटीक FPKM अनुमान प्राप्त करने के लिए कदम 3.2.1 और 3.2.2 में वर्णित के रूप में Tophat और कफ़लिंक का एक दूसरा रन किया जा सकता है.
- आरएनए Seq (से प्राप्त UCSC संस्करण mm9, माउस संदर्भ जीनोम को पढ़ता बनती अंत मानचित्र http://cufflinks.cbcb.umd.edu/igenomes.html का उपयोग करता है) Tophat सॉफ्टवेयर का उपयोग (संस्करण 1.3.3) 27, Bowtie मैपर (संस्करण 0.12.7) 28 पढ़ा. Tophat अभिव्यक्ति के स्तर का आकलन सटीकता में सुधार करने के लिए "-नहीं-उपन्यास-juncs" विकल्प के साथ आपूर्ति की है.
- Differentiall पता लगानेY DESeq पैकेज 30 का उपयोग जीन व्यक्त किया.
- DESeq के इनपुट एक कच्चे पढ़ें मायने रखता तालिका है. इस तरह के एक मेज प्राप्त करने के लिए, HTSeq वेबसाइट से डाउनलोड किया जा सकता है जो HTSeq अजगर पैकेज के साथ वितरित htseq गिनती स्क्रिप्ट का उपयोग ( http://www-huber.embl.de/users/anders/HTSeq/doc/count.html ) .
- Samtools, अजगर, और htseq गिनती programsare सिस्टम में स्थापित सुनिश्चित करें. चित्रा S4A में आदेश का उपयोग कर Tophat उत्पादन से कच्चे पढ़ें गिनती संख्या प्राप्त करते हैं.
- 'Raw_Count_Table.txt' एक्सेल का उपयोग, 'ExperimentDesign.txt' फाइल तैयार. कॉपी और DESeq आर पैकेज (चित्रा S4B) के लिए .txt प्रारूप में सामग्री को बचाने के.
- प्रणाली में अनुसंधान कार्यक्रम को स्थापित करें. टर्मिनल में, 'आर' और प्रेस ENTER.A स्क्रीन संदेश जाएगा appearas चित्रा S4C में दिखाया.
- 'Raw_C पढ़ेंचित्रा S4D में आदेश का उपयोग कर अनुसंधान में ount_Table.txt ',' ExperimentDesign.txt '.
- चित्रा S4E में आदेश का उपयोग DESeq पैकेज लोड करें.
- आर (चित्रा S4F) में खंड करना शर्तों.
- सामान्यीकृत गणना मेज पर नकारात्मक द्विपदिक परीक्षण चलाने के लिए चित्रा S4G में आदेश का उपयोग करें.
- एक .csv फ़ाइल में उत्पादन महत्वपूर्ण अंतर व्यक्त जीनों को चित्रा S4H में आदेश का उपयोग करें.
- DESeq के इनपुट एक कच्चे पढ़ें मायने रखता तालिका है. इस तरह के एक मेज प्राप्त करने के लिए, HTSeq वेबसाइट से डाउनलोड किया जा सकता है जो HTSeq अजगर पैकेज के साथ वितरित htseq गिनती स्क्रिप्ट का उपयोग ( http://www-huber.embl.de/users/anders/HTSeq/doc/count.html ) .
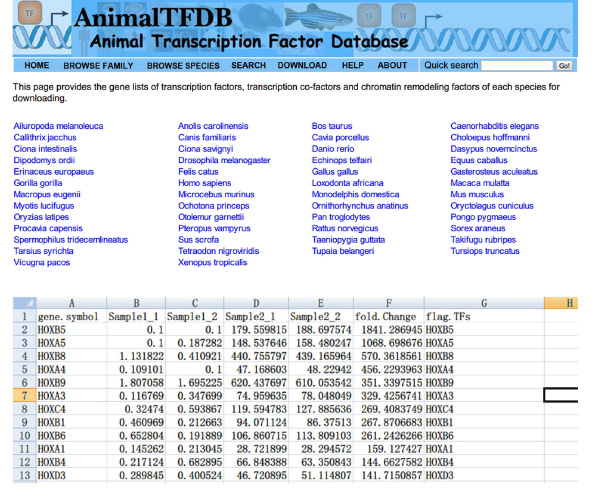
- एक्सेल का उपयोग कर नमूने भर में देखने का प्रतिलेखन कारक '(TFS) FPKM मूल्यों. एक दूसरे को काटना डे जीन मेज और TFS मेज. जीन विभिन्न प्रतिलेखन कारक व्यक्त कर रहे हैं दोनों तालिका के हैं.
- वेबसाइट पर जाएँ http://www.bioguo.org/AnimalTFDB/download.php और प्रतिलेखन कारक डाउनलोड. फिर <(एक्सेल में डे प्रतिलेखन कारक खोज मजबूत> चित्रा S5).
- UCSC जीनोम ब्राउज़र दृश्य के लिए .bigwig फ़ाइल सृजन.
- वेबसाइट से 'bedtools' सॉफ्टवेयर पैकेज डाउनलोड https://github.com/arq5x/bedtools2 और प्रणाली 31 में सॉफ्टवेयर स्थापित करें. वेबसाइट से UCSC उपकरण 'bedGraphToBigWig' डाउनलोड http://hgdownload.cse.ucsc.edu/admin/exe/ और सिस्टम में सॉफ्टवेयर स्थापित करें.
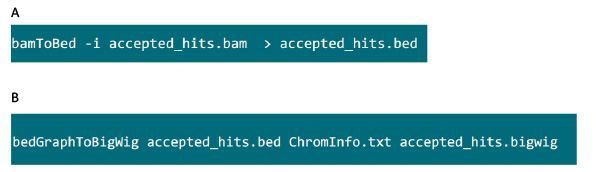
- .bam फ़ाइल वाले फ़ोल्डर में, .bed फ़ाइल में Tophat द्वारा उत्पन्न .bam फ़ाइल में परिवर्तित करने के लिए चित्रा S6A में आदेश का उपयोग करें.
- .bed फ़ाइल का उत्पादन किया है, के बाद .bigwig फ़ाइल उत्पन्न करने के लिए चित्रा S6B में आदेश का उपयोग करें. फ़ाइल 'ChromInfo.txt' निम्न URL से प्राप्त किया जा सकता है:arget = "_blank"> http://hgdownload.cse.ucsc.edu/goldenPath/mm9/database/chromInfo.txt.gz.
- UCSC जीनोम ब्राउज़र पर एक कस्टम ट्रैक का निरीक्षण करें. वेबसाइट को देखें http://genome.ucsc.edu/goldenPath/help/customTrack.html UCSC जीनोम ब्राउज़र का उपयोग कर एक कस्टम ट्रैक प्रदर्शित करने के लिए कैसे पर.

चित्रा एस 1: .bcl फ़ाइल परिवर्तित casava सॉफ्टवेयर का उपयोग कर फ़ाइल .fastq लिए.

चित्रा S2: मैपिंग Tophat का उपयोग जीनोम संदर्भ के लिए पढ़ता है.
चित्रा S3: उपन्यास टेप और अभिव्यक्ति के स्तर आकलन की जांच.

चित्रा एस 4: पैकेज DESeq का उपयोग अंतर व्यक्त जीन कॉलिंग.

चित्रा S5: विभिन्न व्यक्त प्रतिलेखन कारक की पहचान.

चित्रा S6: डेटा दृश्य के लिए मानचित्रण परिणाम परिवर्तित.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
लिन-CD34 + और लिन-CD34- EML कोशिकाओं में विभिन्न व्यक्त जीनों का विश्लेषण करने के लिए, हम शाही सेना Seq तकनीक का इस्तेमाल किया. चित्रा 1 प्रक्रियाओं की कार्यप्रवाह से पता चलता है. चुंबकीय सेल छँटाई द्वारा वंश नकारात्मक कोशिकाओं के अलगाव के बाद, हम लिन-एससीए + CD34 + और FACS Aria का उपयोग लिन-एससीए-CD34- कोशिकाओं अलग कर दिया. लिन समृद्ध EML कोशिकाओं विरोधी CD34, विरोधी Sca1 और वंश कॉकटेल एंटीबॉडी के साथ दाग रहे थे. केवल Lin- कोशिकाओं Sca1 और CD34 अभिव्यक्ति के विश्लेषण के लिए gated थे. दो आबादी (एससीए + CD34 + और एससीए-CD34- EML कोशिकाओं) FACS विश्लेषण (चित्रा 2) 6 से मनाया जा सकता है.
सेल जुदाई के बाद, हम क्रमशः CD34 + और CD34- कोशिकाओं से कुल शाही सेना निकाले और शाही सेना की गुणवत्ता का विश्लेषण किया. आरएनए Seq डेटा की सटीकता काफी हद तक शाही सेना Seq पुस्तकालय की गुणवत्ता पर निर्भर करता है और कुल शाही सेना की गुणवत्ता एक उच्च गुणवत्ता वाले पुस्तकालय की तैयारी के लिए महत्वपूर्ण है. उच्च गुणवत्ता आरएनए नमूना 1 के बीच एक आयुध डिपो 260/280 मान होना चाहिए.8 और 2.0. स्पेक्ट्रोफोटोमीटर का उपयोग करने के अलावा, आरएनए गुणवत्ता आगे Bioanalyzer से अधिक सटीकता के साथ मूल्यांकन किया गया था. 3 9.4 के बराबर Rin के साथ एक उच्च गुणवत्ता शाही सेना के नमूने के परिणाम से पता चलता है. RIN के मूल्य से अधिक 9 के साथ ही उच्च गुणवत्ता वाले कुल शाही सेना नमूना mRNA के निष्कर्षण और बाद में पुस्तकालय निर्माण प्रक्रियाओं के लिए इस्तेमाल किया गया था.
Ribosomal शाही सेना सेल में शाही सेना के सबसे प्रचुर प्रकार है. वर्तमान में दो मुख्य रणनीतियों, rRNA की कमी या polyadenylated mRNA (पाली-ए mRNA) के सकारात्मक चयन, पुस्तकालय निर्माण से पहले लक्ष्य शाही सेना के संवर्धन के लिए उपयोग किया जाता है. गैर polyadenylated आरएनए प्रजातियों पाली-ए mRNA की चयन दौरान खो रहे हैं. इसके विपरीत, इस तरह के RiboMinus रूप rRNA कमी तरीकों गैर polyadenylated आरएनए प्रजातियों की रक्षा कर सकता है. हमारे अध्ययन का उद्देश्य विभिन्न प्रकार हम पुस्तकालय constru से पहले लक्ष्य RNAs के संवर्धन के लिए पाली-ए mRNA के चयन पद्धति का इस्तेमाल किया, दो प्रकार की कोशिकाओं में जीन कोडिंग व्यक्त देखने के लिए हैction है. पुस्तकालय निर्माण समाप्त हो गया था, पुस्तकालय में डीएनए टुकड़े का आकार Bioanalyzer का उपयोग अनुक्रमण से पहले जाँच की थी. 4 के बारे में 300 बीपी पर टुकड़ा आकार चोटियों के साथ एक अच्छी गुणवत्ता पुस्तकालय से पता चलता है.
बाद के चरण में, पुस्तकालय उच्च throughput अनुक्रमण के अधीन था. सिद्धांत रूप में, लंबे समय तक पढ़ा लंबाई पढ़ें मानचित्रण के लिए मददगार होगा. इसे पढ़ने के कारण डुप्लीकेट जीन या जीन परिवार के सदस्यों के बीच समानता के लिए कई स्थानों के लिए मैप किया गया है कि संभावना को कम कर सकते हैं. जोड़ी के अंत अनुक्रमण दृश्यों टुकड़े के दोनों सिरों से हो रहे हैं, को चुना पढ़ें लंबाई औसत टुकड़े लंबाई की तुलना में आधे से कम होना चाहिए. प्रयोग का मुख्य लक्ष्य के बजाय प्रतिलिपि संरचना के निर्माण की अभिव्यक्ति के स्तर को मापने के लिए है, तो एक-अंत बहुत अधिक जानकारी खोने के बिना लागत कम कर सकते हैं (75 या 100 बीपी) पढ़ा. बनती अंत अनुक्रमण प्रतिलिपि संरचना निर्माण और छोटे के लिए अधिक उपयोगी हैलंबाई पढ़ा लागत को कम करने के लिए इस्तेमाल किया जा सकता है. पर्याप्त धन उपलब्ध होने पर निश्चित रूप से, लंबे समय तक पढ़ा लंबाई पसंद किया जाता है.
अंतर अभिव्यक्ति विश्लेषण के लिए, DESeq के अलावा अन्य कई विकल्प एल्गोरिदम रहे हैं. 32 cuffdiff नामित कफ़लिंक पैकेज में शामिल एक भी नहीं है. DESeq सबसे व्यापक रूप से इस्तेमाल गिनती आधारित डे जीन विश्लेषण एल्गोरिदम में से एक है. नकारात्मक द्विपद वितरण - DESeq विधि एक अच्छी तरह से विशेषता सांख्यिकी मॉडल पर आधारित है. हमारे अनुभव में, DESeq cuffdiff के लिए तुलना में अधिक स्थिर है. Cuffdiff के प्रारंभिक संस्करणों अक्सर डे जीन की काफी अलग नंबर दे. इसलिए हम यहाँ डे विश्लेषण के लिए DESeq इस्तेमाल किया.
प्रतिलेखन कारक सेल भाग्य निर्धारण के लिए महत्वपूर्ण होते हैं, क्योंकि हम काफी विभिन्न व्यक्त प्रतिलेखन पर केंद्रित 33 कारकों. TFS लिन-CD34 + और लिन-CD34- के बीच> 1.5 गुना पाया गया बदल गया है और Figur (हीटमैप पर दिखाए जाते हैंई 5) 2. विशेष रूप से, लिन-CD34 + कोशिकाओं में Tcf7 के रिश्तेदार अभिव्यक्ति के स्तर लिन-CD34- कोशिकाओं में है कि अधिक से अधिक से 100 गुना अधिक है. इस प्रकार Tcf7 EML सेल आत्म नवीकरण और भेदभाव 2 के नियमन में Tcf7 के समारोह पुष्टि करने के लिए आगे चिप अनुक्रमण (chromatin immunoprecipitation और अनुक्रमण) विश्लेषण और कार्यात्मक परीक्षण के लिए चुना गया था.

चित्रा 1:. प्रक्रियाओं के कार्यप्रवाह लिन-CD34 + और लिन-CD34- कोशिकाओं चुंबकीय सेल जुदाई प्रणाली और प्रतिदीप्ति सक्रिय सेल छँटाई विधि से अलग हो गए थे. कुल शाही सेना mRNA के शोधन और पुस्तकालय निर्माण के द्वारा पीछा निकाला गया था. पुस्तकालय गुणवत्ता का विश्लेषण करने के बाद, नमूने उच्च throughput अनुक्रमण के अधीन थे. डेटा का विश्लेषण किया और विभिन्न प्रतिलेखन कारक व्यक्त किया गया पहचान की गई.

चित्रा 2:. लिन-CD34 + और लिन-CD34- EML 6 Lin- EML कोशिकाओं चुंबकीय सेल छँटाई से समृद्ध थे कोशिकाओं के पृथक्करण. Lin- कोशिकाओं विरोधी CD34, विरोधी Sca1 और वंश मिश्रण एंटीबॉडी के साथ दाग रहे थे. Lin- कोशिकाओं CD34 और Sca1 की अभिव्यक्ति के लिए gated थे. लिन-CD34 + एससीए + और लिन-CD34-SCA- EML सेल आबादी हल किया गया.

चित्रा 3:. उच्च गुणवत्ता वाले कुल शाही सेना नमूना का एक प्रतिनिधि कुल शाही सेना की गुणवत्ता Bioanalyzer द्वारा मूल्यांकन किया गया था. शाही सेना वफ़ादारी संख्या 9.4 (फू, प्रतिदीप्ति इकाइयों) है.

चित्रा 4:. बनती अंत पुस्तकालय के टुकड़े आकार रेंज पुस्तकालय का डीएनए आकार वितरण Bioanalyzer उपयोग कर विश्लेषण किया गया था. अधिकांश टुकड़े 250-500 बीपी के आकार सीमा के भीतर हैं.

चित्रा 5:. विभिन्न व्यक्त प्रतिलेखन कारक लिन-CD34 + कोशिकाओं और लिन-CD34- कोशिकाओं के बीच 2 (> 1.5 गुना) प्रत्येक कोशिका प्रकार के लिए, दो स्वतंत्र प्रयोगों प्रदर्शन किया गया. लाल रंग और नीचे विनियमित जीन हरे रंग के रूप में संकेत कर रहे हैं के रूप में विनियमित जीन संकेत कर रहे हैं.
तालिका 1: बफ़र और सेल संस्कृति माध्यमों.
| सॉफ्टवेयर | प्रयोग | संदर्भ | |||
| 1.2.7 bowtie | मानचित्रण के लिए Tophat द्वारा प्रयुक्त | [28] | |||
| Tophat 1.3.3 | मैपिंग संदर्भ जीनोम को वापस पढ़ता | [27] | |||
| कफ़लिंक 1.3.0 | देखिए निर्माण और अभिव्यक्ति के स्तर अनुमान | [29] | |||
| DESeq 1.16.0 | अंतर अभिव्यक्ति विश्लेषण | [30] | Bedtools 2.18 | .bed फ़ाइल में .bam फ़ाइल में परिवर्तित | [31] |
| bedGraphToBigWig | फ़ाइल .bigwig को .bed फ़ाइल में परिवर्तित | http://genome.ucsc.edu/ |
तालिका 2: डेटा विश्लेषण के लिए सॉफ्टवेयर की सूची.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
स्तनधारी transcriptome 34-38 बहुत जटिल है. आरएनए Seq प्रौद्योगिकी यह जीन अभिव्यक्ति विश्लेषण के लिए अन्य तरीकों पर कई फायदे हैं transcriptome विश्लेषण, उपन्यास टेप का पता लगाने और एकल nucleotide भिन्नता खोज आदि के अध्ययन में एक तेजी से महत्वपूर्ण भूमिका निभाता है. शुरूआत में उल्लेख किया है, यह माइक्रोएरे के संकरण कलाकृतियों पर काबू पा और उपन्यास टेप डी नोवो की पहचान करने के लिए इस्तेमाल किया जा सकता है. शाही सेना अनुक्रमण की एक सीमा सेंगर अनुक्रमण की तुलना में अपेक्षाकृत कम पढ़ा लंबाई है. हालांकि, अनुक्रमण प्रौद्योगिकी के तेजी से सुधार के साथ, लंबाई लगातार बढ़ती जा रही है पढ़ें. इस पत्र में, हम माउस EML सेल आत्म नवीकरण और भेदभाव में संभावित कुंजी नियामकों की पहचान करने के लिए इस तकनीक का उपयोग करने की विस्तृत तरीकों प्रदान करते हैं.
इस प्रोटोकॉल के लिए पहला महत्वपूर्ण कदम EML सेल संस्कृति है. EML एक hematopoietic अग्रदूत सेल लाइन है और हालांकि यह हो सकता हैएस सी एफ के साथ बड़ी मात्रा में प्रचारित किया. EML कोशिकाओं का संवर्धन हालत सामान्य अमर सेल लाइनों की तुलना में अधिक ध्यान देने की आवश्यकता. कोशिकाओं खिलाया और कोमल आपरेशन के साथ एक नियमित आधार पर passaged किया जाना चाहिए; अन्यथा कोशिकाओं आत्म नवीकरण और भेदभाव के उनके गुणों में परिवर्तन और कोशिका मृत्यु से गुजरना सकता है. पर्याप्त कोशिकाओं को इकट्ठा करने के बाद पहले कदम के रूप में, हम एक चुंबकीय सक्रिय सेल छँटाई प्रणाली का उपयोग वंश नकारात्मक कोशिकाओं को अलग किया. तो फिर हम प्रतिदीप्ति सक्रिय सेल छँटाई का उपयोग CD34 + और CD34- कोशिकाओं अलग कर दिया. EML कोशिकाओं सामान्य रूप से जुदाई के बाद समान होना चाहिए आरएनए निष्कर्षण और CD34 + के नंबर और CD34- कोशिकाओं के लिए उपयोग करने से पहले कम से कम 10 पीढ़ियों passaged कर रहे हैं. दो आबादी सेल संख्या में काफी भिन्नता है, यह संस्कृति को त्यागने और संस्कृति के लिए सेल के शेयर का एक और ट्यूब फिर से पिघलना करने के लिए सलाह दी जाती है.
CD34 + और CD34- सेल की जुदाई के बाद, कुल शाही सेना निष्कर्षण, इस सेंट के लिए एक और महत्वपूर्ण कदम था प्रदर्शनudy. उच्च गुणवत्ता शाही सेना अनुक्रमण डेटा की सटीकता का वादा किया है जो एक उच्च गुणवत्ता वाले पुस्तकालय, के निर्माण के लिए आधार है. इस महत्वपूर्ण चरण में, RNase साथ किसी भी संपर्क से बचा जाना चाहिए. सभी अभिकर्मकों मुक्त RNase किया जाना चाहिए. यह शाही सेना से निपटने जबकि हर समय दस्ताने पहनने के लिए महत्वपूर्ण है. उच्च गुणवत्ता शाही सेना नमूना 1.8 और 2.0 के बीच एक आयुध डिपो 260/280 मूल्य है. आरएनए युक्त जलीय चरण एकत्रित करते समय, आरएनए नमूना के साथ किसी भी जैविक चरण ले जाने के लिए नहीं सावधान रहना होगा. ऐसे शाही सेना में फिनोल या क्लोरोफॉर्म के रूप में किसी भी अवशिष्ट कार्बनिक सॉल्वैंट्स 1.65 से कम एक OD260 / 280 मूल्य पर नतीजा होगा. OD260 / 280 मूल्य 1.65 से कम है, इथेनॉल के साथ फिर से शाही सेना वेग. 75% इथेनॉल के साथ धोने के बाद, नहीं overdry आरएनए गोली करते हैं. सुखाने आरएनए गोली पूरी तरह से शाही सेना की घुलनशीलता को प्रभावित और शाही सेना की कम पैदावार को बढ़ावा मिलेगा.
इस प्रोटोकॉल के लिए अगले महत्वपूर्ण कदम पुस्तकालय तैयारी है. कुल शाही सेना निष्कर्षण, दूषित डीएनए को हटाने के लिए DNase का उपयोग करने का एक कदम के बाद मैंडीएनए संदूषण इस्तेमाल किया कुल शाही सेना की राशि का गलत आकलन में परिणाम हो सकता है के बाद से अत्यधिक की सिफारिश की. यह करने के बाद लंबी अवधि के भंडारण के बाद, शाही सेना अलगाव के बाद तुरंत नीचे की ओर कार्यविधि को पूरा करने और प्रक्रिया-विगलन फ्रीज करने के लिए सिफारिश की है, आरएनए कुछ हद तक नीचा दिखाना होगा. शाही सेना अलगाव के बाद बाद के चरणों तुरंत नहीं किया जा सकता है, -80 डिग्री सेल्सियस में शाही सेना की दुकान. कुल शाही सेना mRNA के शोधन और सीडीएनए संश्लेषण के लिए प्रयोग किया जाता है इससे पहले, गुणवत्ता हमेशा जाँच की जानी चाहिए. केवल उच्च गुणवत्ता शाही सेना पुस्तकालय तैयारी के लिए इस्तेमाल किया जा सकता है. कम गुणवत्ता या अपमानित शाही सेना का प्रयोग 'समाप्त होता है अति-प्रतिनिधित्व 3 का नेतृत्व करने के लिए हो सकता है. अनुक्रमण पहले, पुस्तकालय गुणवत्ता अधिकतम अनुक्रमण दक्षता सुनिश्चित करने के लिए मूल्यांकन किया गया था.
डेटा विश्लेषण भाग में, एक संदर्भ transcriptome बिना कफ़लिंक की एक रन के प्रदर्शन के बाद, हम दूसरी बार के लिए फाइल और रन Tophat और कफ़लिंक .gtf एक संदर्भ के लिए फार्म ज्ञात टेप के साथ उपन्यास टेप संयुक्त.यह केवल एक बार चल तुलना में ज्यादा सटीक FPKM आकलन उपलब्ध कराने के बाद से यह दो रन की प्रक्रिया, की सिफारिश की है. डेटा विश्लेषण के बाद, विभिन्न व्यक्त जीनों की पहचान की गई. डाउनस्ट्रीम प्रयोगों इन विट्रो में और vivo में जीन के कार्य को मान्य करने के लिए किया जा सकता है. हमारे पिछले प्रकाशन 2 में, हम काफी विभिन्न व्यक्त प्रतिलेखन कारक चुना है और chromatin immunoprecipitation और अनुक्रमण (चिप Seq) प्रदर्शन से इन कारकों के जीनोम बाध्यकारी साइट की पहचान की. इसके अलावा, हम Tcf7 के कार्यात्मक प्रभाव का परीक्षण करने के shRNA पछाड़ना परख आवेदन किया. हम नीचे विनियमित जीन काफी CD34 + कोशिकाओं में समृद्ध हो पाए थे, जबकि Tcf7 पछाड़ना कोशिकाओं में, विनियमित अप जीन अत्यधिक CD34- कोशिकाओं में समृद्ध जीन, पाया गया कि. इसलिए, Tcf7 पछाड़ना कोशिकाओं के जीन अभिव्यक्ति प्रोफाइल एक मॉडल प्रणाली के रूप में EML सेल का उपयोग कर, एक आंशिक रूप से विभेदित CD34- state.Overall की ओर स्थानांतरित कर दियाशाही सेना अनुक्रमण प्रौद्योगिकी और कार्यात्मक assays के साथ युग्मित, हम पहचान और EML सेल आत्म नवीकरण और भेदभाव का एक महत्वपूर्ण नियामक के रूप में Tcf7 की पुष्टि की.
Subscription Required. Please recommend JoVE to your librarian.
Materials
| Name | Company | Catalog Number | Comments |
| Antibiotic-Antimycotic | Invitrogen | 15240-062 | BHK cell culture |
| Anti-Mouse CD34 FITC | eBioscience | 11-0341-81 | FACS sorting |
| Anti-Mouse Ly-6A/E (Sca-1) PE | eBioscience | 12-5981-81 | FACS sorting |
| APC Mouse Lineage Antibody Cocktail | BD Biosciences | 558074 | FACS sorting |
| BD FACSAria Cell Sorter | BD Biosciences | Special offer sysmtem | FACS sorting |
| Corning™ Cell Culture Treated Flasks 75 cm2 | Corning incorporated | 430641 | Cell culture |
| Corning™ Cell Culture Treated Flasks 25 cm2 | Corning incorporated | 430639 | Cell culture |
| Deoxyribonuclease I, Amplification Grade | Invitrogen | 18068-015 | Library preparation |
| DMEM | Invitrogen | 11965-092 | BHK cell culture |
| DPBS | Gibco | 14190 | Cell culture |
| HI FBS | Invitrogen | 16140071 | BHK cell culture |
| Horse Serum | Invitrogen | 16050-122 | EML cell culture |
| IMDM | HyClone | SH30228.02 | EML cell culture |
| L-Glutamine | Invitrogen | 25030-081 | Cell culture |
| Lineage Cell Depletion Kit, mouse | Miltenyi Biotec | 130-090-858 | Isolation of lineage negative cells |
| NanoVue Plus spectrophotometer | GE Healthcare | 28-9569-62 | Quality control |
| Thermo Scientific™ Napco™ 8000 Water-Jacketed CO2 Incubators | Thermo Scientific | 15-497-002 | Cell culture |
| Penicillin-Streptomycin | Invitrogen | 15140-122 | EML cell culture |
| TRIzol® Reagent | Invitrogen | 15596-018 | RNA exraction |
| TruSeq™ RNA Sample Prep Kit v2 -Set B (48 rxn) | Illumina | RS-122-2002 | Library preparation |
| 2100 Electrophoresis Bioanalyzer Instrument | Agilent | G2939AA | Quality control |
| 0.25% Trypsin-EDTA | Gibco | 25200 | Cell culture |
| 0.45 µm Syringe Filters | Nalgene | 190-2545 | Cell culture |
References
- Chambers, S. M., Goodell, M. A. Hematopoietic stem cell aging: wrinkles in stem cell potential. Stem Cell Rev. 3, 201-211 (2007).
- Wu, J. Q., et al. Tcf7 is an important regulator of the switch of self-renewal and differentiation in a multipotential hematopoietic cell line. PLoS genetics. 8, (2012).
- Ye, Z. J., et al. Complex interactions in EML cell stimulation by stem cell factor and IL-3. Proceedings of the National Academy of Sciences of the United States of America. 108, 4882-4887 (2011).
- Tsai, S., Bartelmez, S., Sitnicka, E., Collins, S. Lymphohematopoietic progenitors immortalized by a retroviral vector harboring a dominant-negative retinoic acid receptor can recapitulate lymphoid, myeloid, and erythroid development. Genes Dev. 8, 2831-2841 (1994).
- Weiler, S. R., et al. D3: a gene induced during myeloid cell differentiation of Linlo c-Kit+ Sca-1(+) progenitor cells. Blood. 93, 527-536 (1999).
- Ye, Z. J., Kluger, Y., Lian, Z., Weissman, S. M. Two types of precursor cells in a multipotential hematopoietic cell line. Proc Natl Acad Sci U S A. 102, 18461-18466 (2005).
- Wang, Z., Gerstein, M., Snyder, M. RNA-Seq: a revolutionary tool for transcriptomics. Nature reviews. Genetics. 10, 57-63 (2009).
- Chu, Y., Corey, D. R. RNA sequencing: platform selection, experimental design, and data interpretation. Nucleic acid therapeutics. 22, 271-274 (2012).
- Hornett, E. A., Wheat, C. W. Quantitative RNA-Seq analysis in non-model species: assessing transcriptome assemblies as a scaffold and the utility of evolutionary divergent genomic reference species. BMC genomics. 13, 361 (2012).
- Eswaran, J., et al. RNA sequencing of cancer reveals novel splicing alterations. Scientific reports. 3, 1689 (2013).
- Wang, E. T., et al. Alternative isoform regulation in human tissue transcriptomes. Nature. 456, 470-476 (2008).
- Wu, J. Q., et al. Dynamic transcriptomes during neural differentiation of human embryonic stem cells revealed by short, long, and paired-end sequencing. Proceedings of the National Academy of Sciences of the United States of America. 107, 5254-5259 (2010).
- Loraine, A. E., McCormick, S., Estrada, A., Patel, K., Qin, P. RNA-seq of Arabidopsis pollen uncovers novel transcription and alternative splicing. Plant physiology. 162, 1092-1109 (2013).
- Edgren, H., et al. Identification of fusion genes in breast cancer by paired-end RNA-sequencing. Genome biology. 12, 6 (2011).
- Ilott, N. E., Ponting, C. P. Predicting long non-coding RNAs using RNA sequencing. Methods. 63, 50-59 (2013).
- Sun, L., et al. Prediction of novel long non-coding RNAs based on RNA-Seq data of mouse Klf1 knockout study. BMC bioinformatics. 13, 331 (2012).
- Luo, S. MicroRNA expression analysis using the Illumina microRNA-Seq Platform. Methods in molecular biology. 822, 183-188 (2012).
- Bolduc, F., Hoareau, C., St-Pierre, P., Perreault, J. P. In-depth sequencing of the siRNAs associated with peach latent mosaic viroid infection. BMC molecular biology. 11, 16 (2010).
- Chepelev, I., Wei, G., Tang, Q., Zhao, K. Detection of single nucleotide variations in expressed exons of the human genome using RNA-Seq. Nucleic acids research. 37, 106 (2009).
- Djari, A., et al. Gene-based single nucleotide polymorphism discovery in bovine muscle using next-generation transcriptomic sequencing. BMC genomics. 14, 307 (2013).
- Murphy, D. Gene expression studies using microarrays: principles, problems, and prospects. Advances in physiology education. 26, 256-270 (2002).
- Chen, K., et al. RNA-seq characterization of spinal cord injury transcriptome in acute/subacute phases: a resource for understanding the pathology at the systems level. PLoS one. 8, 72567 (2013).
- Marioni, J. C., Mason, C. E., Mane, S. M., Stephens, M., Gilad, Y. RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome research. 18, 1509-1517 (2008).
- Ramskold, D., Kavak, E., Sandberg, R. How to analyze gene expression using RNA-sequencing data. Methods in molecular biology. 802, 259-274 (2012).
- Glenn, T. C. Field guide to next-generation DNA sequencers. Mol Ecol Resour. 11, 759-769 (2011).
- Trapnell, C., et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nature protocols. 7, 562-578 (2012).
- Trapnell, C., Pachter, L., Salzberg, S. L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 25, 1105-1111 (2009).
- Langmead, B., Trapnell, C., Pop, M., Salzberg, S. L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome biology. 10, 25 (2009).
- Trapnell, C., et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature. 28, 511-515 (2010).
- Anders, S., Huber, W. Differential expression analysis for sequence count data. Genome biology. 11, 106 (2010).
- Quinlan, A. R., Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 26, 841-842 (2010).
- Cheranova, D., et al. RNA-seq analysis of transcriptomes in thrombin-treated and control human pulmonary microvascular endothelial cells. J Vis Exp. , (2013).
- Zhang, H. M., et al. AnimalTFDB: a comprehensive animal transcription factor database. Nucleic acids research. 40, 144-149 (2012).
- Wu, J. Q., et al. Systematic analysis of transcribed loci in ENCODE regions using RACE sequencing reveals extensive transcription in the human genome. Genome Biol. 9, 3 (2008).
- Wu, J. Q., et al. Large-scale RT-PCR recovery of full-length cDNA clones. Biotechniques. 36, 690-696 (2004).
- Wu, J. Q., Shteynberg, D., Arumugam, M., Gibbs, R. A., Brent, M. R. Identification of rat genes by TWINSCAN gene prediction, RT-PCR, and direct sequencing. Genome Res. 14, 665-671 (2004).
- Dewey, C., et al. Accurate identification of novel human genes through simultaneous gene prediction in human, mouse, and rat. Genome Res. 14, 661-664 (2004).
- Wu, J. Characterize Mammalian Transcriptome Complexity. , LAP Lambert Academic Publishing. (2011).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}