

Summary
RNA-Sequenzierung und Bioinformatik-Analysen wurden verwendet, um deutlich und differentiell exprimiert Transkriptionsfaktoren in Lin-CD34 + und Lin-CD34- Subpopulationen von Maus EMLcells identifizieren. Diese Transkriptionsfaktoren könnten eine wichtige Rolle bei der Bestimmung der Wechsel zwischen Selbsterneuerung Lin-CD34 + und teilweise differenziert Lin-CD34- Zellen spielen.
Abstract
Hämatopoetischen Stammzellen (HSCs) werden klinisch zur Behandlung Transplantation verwendet, um blutbildende System des Patienten in vielen Krankheiten, wie Leukämie und Lymphom aufzubauen. Aufklärung der Mechanismen, die HSK Selbsterneuerung und Differenzierung ist wichtig, für die Anwendung der HSK für Forschung und klinische Anwendungen. Jedoch ist es nicht möglich, große Mengen von HSZ erhalten aufgrund ihrer Unfähigkeit, in vitro zu proliferieren. Um diese Hürde zu überwinden, verwendeten wir eine Knochenmark der Maus abgeleitete Zelllinie, die EML (Erythroid, myeloische und lymphatische) Zelllinie, als Modellsystem für diese Studie.
RNA-Sequenzierung (RNA-Seq) wurde zunehmend zur Microarray für Genexpressionsstudien zu ersetzen. Wir berichten hier über eine detaillierte Verfahren zur Verwendung von RNA-Seq-Technologie, um die potenziellen Schlüsselfaktoren bei der Regulation der EML Zellselbsterneuerung und Differenzierung zu untersuchen. Die in diesem Papier Protokoll ist in drei Teile unterteilt. Das erste Part erklärt, wie man Kultur EML Zellen und separaten Lin-CD34 + und Lin-CD34- Zellen. Der zweite Teil des Protokolls bietet detaillierte Verfahren für Gesamt-RNA Vorbereitung und die anschließende Konstruktion der Bibliothek für Hochdurchsatz-Sequenzierung. Der letzte Teil beschreibt die Methode für die RNA-Seq Datenanalyse und erklärt, wie die Daten verwenden, um differentiell exprimierte Transkriptionsfaktoren zwischen Lin-CD34 + und Lin-CD34- Zellen zu identifizieren. Die signifikant differentiell exprimiert Transkriptionsfaktoren wurden identifiziert, um die potenziellen Schlüsselregulatoren steuern EML Zellselbsterneuerung und Differenzierung. Im Diskussionsteil dieser Arbeit unterstreichen wir die wichtigsten Schritte für die erfolgreiche Entwicklung dieses Experiments.
Dieses Papier Zusammengefasst bietet ein Verfahren zur Verwendung von RNA-Seq-Technologie, um potentielle Regulatoren der Selbsterneuerung und Differenzierung in EML-Zellen zu identifizieren. Die Schlüsselfaktoren identifiziert werden, um nachgeschaltete Funktionsanalyse in vitro und i unterzogenn vivo.
Introduction
Hämatopoetische Stammzellen sind selten Blutzellen, die vor allem in der Knochenmark Erwachsener Nische befinden. Sie sind für die Produktion von Zellen erforderlich ist, um das Blut wieder aufzufüllen und das Immunsystem 1 verantwortlich. Als eine Art von Stammzellen, HSK sind in der Lage, sowohl die Selbsterneuerung und Differenzierung. Erforschung von Prozessen, die das Schicksal Entscheidung des HSK zu steuern, entweder in Richtung Selbsterneuerung oder Differenzierung, werden wertvolle Hinweise auf die Manipulation von HSCs für Blutkrankheit Forschungen und klinische Nutzung 2 bieten. Ein Problem von den Forschern konfrontiert ist, dass HSCs aufrechterhalten und in vitro in einem sehr begrenzten Ausmaß erweitert werden; die überwiegende Mehrheit ihrer Nachkommen sind teilweise in der Kultur 2 differenziert.
Um Schlüsselregulatoren, die die Prozesse der Selbsterneuerung und Differenzierung in einer genomweiten Maßstab steuern zu identifizieren, haben wir eine Maus primitiven hämatopoetischen Vorläuferzelllinie EML als Modellsystem. Thwird Zelllinie wurde von murinen Knochenmark 3,4 abgeleitet. Wenn mit verschiedenen Wachstumsfaktoren zugeführt wird, kann EML Zellen in erythroiden, myeloiden und lymphoiden Zellen in vitro 5 unterscheiden. Wichtig ist, dass diese Zelllinie in großer Menge in Kulturmedium, das Stammzellfaktor (SCF) und Beibehaltung ihrer multipotentiality vermehrt werden. EML Zellen in Subpopulationen von sich selbst erneuernden Lin-SCA + CD34 + getrennt und teilweise differenziert Lin-SCA-CD34- Zellen auf Oberflächenmarker CD34 und SCA 6. Ähnlich wie bei kurzfristigen HSCs, SCA + CD34 + Zellen sind in der Lage sich selbst zu erneuern. Wenn sie mit SCF, Lin-SCA + CD34 + Zellen, behandelt schnell regenerieren einer gemischten Population von Lin-SCA + CD34 + und Lin-SCA-CD34- Zellen und weiter zu vermehren 6. Die beiden Populationen ähnliche Morphologie und haben ähnliche Niveaus der c-kit-mRNA und Protein-6. Lin-SCA-CD34- Zellen können Vermehrungsmedium, das IL-3 anstelle von SCF 3. Unveiling die Schlüsselregulatoren in der EML Zellschicksal Entscheidung besseres Verständnis der zellulären und molekularen Mechanismen in frühen Entwicklungs Übergang während der Hämatopoese bieten.
Um die zugrunde liegenden molekularen Unterschiede zwischen den sich selbst erneuernden Lin-SCA + CD34 + und teilweise differenziert Lin-SCA-CD34- Zellen zu untersuchen, RNA-Seq wir differentiell exprimierte Gene zu identifizieren. Insbesondere konzentrieren wir uns auf Transkriptionsfaktoren, wie Transkriptionsfaktoren sind entscheidend bei der Bestimmung des Zellschicksals. RNA-Seq ist eine kürzlich entwickelte Ansatz, der die Fähigkeiten der nächsten Generation Sequenzierung verwendet (NGS) -Technologien zum Profil und zu quantifizieren RNAs aus Genom 7,8 transkribiert. Kurz, Gesamt-RNA poly-A ausgewählt und fragmentiert wie die anfängliche template.The RNA-Matrize wird dann in cDNA unter Verwendung von reverser Transkriptase umgewandelt. Um Volllängen-RNA-Transkripte der Karte unter Verwendung intakter, nicht abgebauter RNA zur Konstruktion von cDNA-Bibliothek ist wichtig. Für die purHaltung der Sequenzierung werden spezifische Adaptersequenzen an beiden Enden der cDNA addiert. Dann wird in den meisten Fällen, cDNA-Moleküle werden durch PCR amplifiziert und in einem Hochdurchsatz Weise sequenziert.
Nach der Sequenzierung, liest der resultiert, kann zu einem Referenzgenom und Transkriptom-Datenbank ausgerichtet werden. Die Anzahl der Lesevorgänge, die Karte an die Referenz-Gen wird gezählt, und diese Information kann verwendet werden, um die Genexpression Pegel abzuschätzen. Das liest kann auch zusammengebaut werden de novo ohne Referenzgenom, so dass das Studium der Transkriptomen in Nicht-Modellorganismen 9. RNA-seq Technik wurde auch verwendet, um Spleiß-Isoformen 10-12, neuer Transkripte 13 und Genfusionen 14 erfassen. Zusätzlich zum Nachweis von Protein-codierenden Genen kann RNA-Seq auch verwendet, um neuartige erfassen und zu analysieren Transkriptionsniveau des nicht-kodierenden RNAs, wie lange werden nicht-kodierende RNA 15,16, MikroRNA 17, 18 usw. siRNA. Wegen tEr Genauigkeit dieser Methode wurde zum Nachweis von single nucleotide Variationen 19,20 verwendet.
Vor dem Aufkommen der RNA-Seq-Technologie, Mikroarray war die wichtigste Methode für die Analyse von Genexpressionsprofil verwendet. Vorgefertigte Sonden synthetisiert und anschließend an einer festen Oberfläche befestigt ist, um einen Mikroarray-Objektträger 21 zu bilden. mRNA extrahiert und in cDNA umgewandelt. Während der reversen Transkription Verfahren werden fluoreszenzmarkierte Nukleotide in die cDNA eingebaut, und die cDNA kann auf die Mikroarray-Objektträger hybridisiert werden. Die Intensität des von einer bestimmten Stelle gesammelt Signals hängt von der Menge an cDNA-Bindung an die spezifische Sonde an dieser Stelle 21. Verglichen mit RNA-Seq-Technologie, hat Microarray mehrere Einschränkungen. Zunächst setzt Microarray auf dem bereits bestehenden Wissen der Gen-Annotation, während RNA-Seq-Technologie ist in der Lage, neue Transkripte bei relativen hohen Hintergrundkonzentration, die seine Verwendung, wenn begrenzt erkennen gene Expressionsniveau ist niedrig. Außerdem hat das RNA-Seq-Technologie viel höheren Dynamikbereich der Erfassungs (8000-fach) 7, wobei aufgrund der Hinter und Sättigung von Signalen, ist die Genauigkeit der Mikroarray sowohl von hoch und niedrig exprimierten Genen 7,22 begrenzt. Schließlich unterscheiden sich Mikroarray-Sonden in ihren Hybridisierungseffizienzen, die bei einem Vergleich relativen Expressionslevel der verschiedenen Transkripte innerhalb einer Probe 23 die Ergebnisse weniger zuverlässig machen. Obwohl RNA-Seq hat viele Vorteile gegenüber Mikroarray ist ihre komplexe Datenanalyse. Dies ist einer der Gründe, warum viele Forscher Mikroarray verwenden noch immer statt RNA-Seq. Verschiedene Methoden der Bioinformatik für RNA-Seq Datenverarbeitung und -analyse 24 erforderlich.
Unter mehreren Next-Generation-Sequencing (NGS) Plattformen, 454, sind Illumina, solide und Ion Torrent die am häufigsten benutzten. 454 war der erste kommerzielle NGS-Plattform. Im Gegensatz zu den anderen Sequenzierplattformenwie Illumina und SOLID erzeugt der 454-Plattform mehr gelesen Länge (durchschnittlich 700 Basis liest) 25. Länger liest sich besser für erstmalige Beschreibung transcriptiome aufgrund ihrer höheren Effizienz montieren 25. Der Hauptnachteil des 454-Plattform ist die hohe Kosten pro Megabasen der Sequenz. Die Illumina und SOLID-Plattformen erzeugen liest mit erhöhten Zahlen und kurze Längen. Die Kosten pro Megabasen-Sequenz ist viel niedriger als die Plattform 454. Aufgrund der großen Zahl von kurzen liest die Illumina und SOLID-Plattformen ist die Datenanalyse viel rechenintensiver. Der Preis des Gerätes und Reagenzien für die Sequenzierung der Ion Torrent Plattform ist billiger und die Sequenzierung Zeit kürzer 25. Allerdings ist die Fehlerrate und die Kosten pro Megabasen der Sequenz sind höher als in der Illumina und SOLID Plattformen. Unterschiedliche Plattformen haben ihre eigenen Vor- und Nachteile und erfordern unterschiedliche Methoden zur Datenanalyse. Die plaTForm sollten auf der Grundlage der Sequenzierung Zweck und der Verfügbarkeit der Mittel gewählt werden.
In diesem Beitrag nehmen wir Illumina RNA-Seq-Plattform als Beispiel. Wir verwendeten EML Zelle als Modellsystem, um die Schlüsselregulatoren in EML Zellselbsterneuerung und Differenzierung zu untersuchen, und eine detaillierte Methoden der RNA-Seq Bibliothekskonstruktion und Datenanalyse für Expressionsniveau Berechnung und neuartige Transkript Erkennung. Wir haben in unserer früheren Veröffentlichung, dass RNA-seq Studie in EML Modellsystem 2, wenn sie mit Funktionsprüfung (zB shRNA Zuschlags) bieten eine leistungsfähige Ansatz zum Verständnis des molekularen Mechanismus der frühen Stadien von hämatopoetischen Differenzierung gezeigt und kann als dienen Modell für die Analyse der Zellselbsterneuerung und Differenzierung im Allgemeinen.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. EML Zellkultur und Trennung von Lin-CD34 + und Lin-CD34- Zellen unter Verwendung Magnetic Cell Sorting System und fluoreszenzaktivierte Zellsortierverfahren
- Herstellung von Babyhamster-Nieren (BHK) Zellkulturmedium für die Stammzellfaktor Sammlung:
- Kultur BHK-Zellen in DMEM-Medium mit 10% FBS in 25 cm 2 -Kolben (Tabelle 1) bei 37 ° C, 5% CO 2 in einem Zellkulturbrutschrank.
- Wenn Zellen wachsen, um 80 bis 90% Konfluenz, waschen Sie die Zellen einmal mit 10 ml PBS. 5 ml 0,25% Trypsin-EDTA-Lösung zu der Monoschicht und Inkubieren der Zellen für 1-5 min bei Raumtemperatur (RT) bis die Zellen abgelöst werden.
- Pipette die Lösung oben und unten vorsichtig aufzubrechen Zellklumpen. 5 ml komplettem DMEM in den Kolben zu Trypsinaktivität stoppen. Sammeln Zellen durch Zentrifugation bei 200 xg für 5 Minuten bei RT.
- Entfernen des Mediums und Resuspendieren des Zellpellets in 10 ml frische BHK-Zellkulturmedium. Übertragungs 2 ml der Zellsuspension aus Schritt 1.1.4 auf eine neue 75 cm 2 eingewogen und 48 ml der frischen BHK-Zellkulturmedium in den Kolben.
- Kultur die BHK-Zellen für zwei Tage zu sammeln und das Kulturmedium. Gang das Medium durch einen 0,45 um-Filter. Bewahren Sie das Medium, in -20 ° C bis zur weiteren Verwendung.
- EML Zellkultur:
- Kultur EML-Zellen (in Suspension) in Grundmedium, das EML BHK-Zellkulturmedium (Tabelle 1) bei 37 ° C, 5% CO 2 in einem Zellkulturbrutschrank.
- Aufrechterhaltung der EML Zellen bei geringer Zelldichte (0,5-5 x 10 5 Zellen / ml) mit dem Spitzendichte von weniger als 6 x 10 5 Zellen / ml. Aufgeteilt, die Zellen alle 2-3 Tage in einem Verhältnis von 1: 5. Passage EML Zellen vorsichtig und entsorgen Sie die Kultur nach Passagieren für 10 Generationen.
- Depletion von Lineage-positiven Zellen:
- Ernten Sie die EML Zellen durch Zentrifugation bei 200 × g for 5 min und waschen Sie die Zellen einmal mit PBS. Sammeln der Zellen durch Zentrifugation bei 200 × g für 5 min.
- Resuspendieren der Zellen mit PBS und zähle die Zellen mit einem Hämozytometer. Bestimmen Sie die Antikörper-Konzentration in der anschließenden Zellseparationsschritt nach der Anzahl der Zellen (beachten Sie bitte die Anweisungen des Anbieters des Zellisolationssystem angeboten beziehen).
- Isolieren Sie die Linie negativ (Lin-) Zellen unter Verwendung Linie Antikörper-Cocktail (Cocktail aus Biotin-konjugierten monoklonalen Antikörper CD5, CD45R (B220), CD11b, Anti-Gr-1 (Ly-6G / C), 7-4 und Ter-119 ) und eine magnetische Zellsortierung System nach den Anweisungen des Herstellers.
- Trennung von Lin-CD34 + und Lin-CD34- Zellen:
- Spin-down der Lin- Zellen aus Schritt 1.3.3 bei 200 × g für 5 min. Zellpellet mit PBS und zählen Sie die Zellen mit einer Zählkammer.
- Waschen der Zellen zweimal mit FACS-Puffer und Pelletierung der Zellen bei 200 xgfür 5 min.
- Beschriften fünf 1,5 ml Mikrozentrifugenröhrchen mit der Nummer 1, 2, 3, 4, 5 auf. Resuspendieren der Zellen mit 100 & mgr; l FACS-Puffer pro 10 6 Zellen (10 6 Zellen pro Röhrchen).
- In 1 ug Anti-Maus-CD34 FITC-Antikörper in Röhrchen 1 und Rohr 2 und die Röhrchen vorsichtig mischen.
- Alle Röhrchen bei 4 ° C inkubieren für 1 h in der Dunkelheit.
- In 0,25 ug PE-konjugierten Anti-Sca1 Antikörper und 20 ul APC-konjugierten Lineage Cocktail Antikörper in Röhrchen 1, 0,25 ug PE-konjugierten Anti-Sca1 Antikörper gegen Rohr 3 und 20 ul APC-konjugierten Lineage Cocktail Antikörpern gegen Rohr 4.
- Mischen Sie alle Röhrchen vorsichtig und Inkubation der Zellen bei 4 ° C für weitere 30 min im Dunkeln.
- Hinzufügen von 300 & mgr; l FACS-Puffer zu den Zellen und Spin-down werden die Zellen bei 200 g für 5 min.
- Mit 500 & mgr; l FACS-Puffer dreimal Waschen der Zellen.
- Zellpellet in 500 ul FACS buffer.
- Verwenden Sie die Zellen in Röhrchen 2, 3, 4 und 5 für die Einrichtung Entschädigung. Isolieren Lin-SCA + CD34 + und Lin-SCA-CD34- Zellen in Rohr 1 mittels FACS Aria.
2. RNA Präparation und Library Construction für Hochdurchsatz-Sequenzierung
- Isolation, Qualitätsanalyse und Quantifizierung von RNA:
- Auszug Gesamt-RNA aus Lin-CD34 + und Lin-CD34- Zellen jeweils mit TRIzol nach fertigt 'Protokoll.
- Entfernen Sie die kontaminierten DNA unter Verwendung Desoxyribonuclease I (DNase I) folgende Protokoll des Herstellers. Gegebenenfalls, speichern die RNA bei -80 ° C bei diesem Schritt für die weitere Verwendung.
- Bewerten Sie die Qualität der Gesamt-RNA mit Bioanalyzer gemäß den Anweisungen des Lieferanten angeboten. Verwenden RNA-Probe mit RNA Integrity Number (RIN) lager als 9.
- Library Construction und Hochdurchsatz-Sequenzierung:
HINWEIS: Dieses Protokoll beschreibt RNA-Seq mit Illumina-Plattform. Fürandere Sequenzierungsplattformen werden verschiedene Bibliothekspräparationsmethoden erforderlich.- Verwenden 0,1-4 ug hochwertige Gesamt-RNA pro Probe für Bibliotheks Vorbereitung. Regel 2 ug Gesamt-RNA aus 10 5 EML Zellen extrahiert werden.
- Verwenden einer RNA-Sequenzierungsprobenvorbereitungssystem für RNA-Reinigung und Fragmentierung, ersten und zweiten Strang der cDNA-Synthese Ende Reparatur, 3'-Enden Adenylierungs, Adapter-Ligation und PCR-Amplifikation, nach den Standardverfahren von detaillierten Instruktionen des Anbieters.
- Günstig wählen PolyA-mRNA mit Oligo-dT-magnetischen Kügelchen und Zersplitterung des mRNA.
- Zuführen reverse Transkription unter Verwendung von Zufallsprimern, um die cDNA zu erhalten, und anschließend die Synthese des zweiten Strangs der cDNA, um doppelsträngige cDNA zu erzeugen.
- Entfernen Sie die 3'-Überhänge und die 5 zu füllen 'Überhänge durch DNA-Polymerase. Adenylat 3'-Enden von cDNA-Fragmenten aus Ligation miteinander zu verhindern.
- Hinzufügen Multiplexschalt Adapter an beide Enden der dscDNA. Zuführen PCR zur Anreicherung von DNA-Fragmenten.
- Messung der A260 / A280, um Informationen über die Konzentration der Bibliothek unter Verwendung eines Spektrophotometers erhalten.
- Beurteilen Sie die Bibliothek Qualität und messen den Größenbereich von DNA-Fragmenten mit einem Bioanalyzer.
3. Datenanalyse
Als Referenz der Software in diesem Teil verwendet wird, finden Sie in (Tabelle 2).
- Datendateiverarbeitung für Downstream-Analyse:
- Konvertieren .bcl (Basis Call-Datei) Datei zu Datei .fastq Verwendung casava Software (Illumina, Version 1.8.2).
- Starten Sie den 'Terminal' im Linux-System. Gehen Sie auf die Datenordner, die die Datendatei von einem Illumina HiSeq2000 Sequenzierungsmaschine enthält. Angenommen das Ergebnis Ordner 'NASboy1 / JiaqianLabData / HiSeq_RUN / 2013_07_11 / 130627_SN860_0309_A_2013-166_H0PW9ADXX /', Typim Befehl in Abbildung S1A, und geben Sie den Datenordner.
- Installieren casava 1.8.2 in das Linux-System. Angenommen, der output ist 'Unaligned', verwenden Sie den Befehl in Abbildung S1B, die Konfigurationsdatei für die Umwandlung vorzubereiten. Verwenden Sie die Option --fastq-Cluster-count 0, um sicherzustellen, nur eine .fastq Datei wird für jede Probe erstellt. Die erzeugte .fastq Datei im .gz-Format. Entpacken Sie es für Downstream-Analyse (Abbildung S1B).
- Nachdem der 'Unaligned' Ordner erzeugt wurde, gehen Sie zum 'Unaligned' Ordner (Abbildung S1C).
- Verwenden Sie den Befehl in Abbildung S1D um den Konvertierungsprozess zu beginnen. Der Parameter '-j' liefert die CPU-Nummer, die verwendet werden.
- Nachdem das System abgeschlossen Umwandlungsprozess, gehen Sie zu dem Ergebnis, Ordner unter 'Unaligned' Ordner (Abbildung S1E).
- Verwenden Sie den Befehl in Abbildung S1F </ Strong>, um die Datei in .fastq.gz .fastq Datei unter jeder Probe Ordner entpacken.
- Konvertieren .bcl (Basis Call-Datei) Datei zu Datei .fastq Verwendung casava Software (Illumina, Version 1.8.2).
- Detect neuer Transkripte und bewerten das Expressionsniveau mit Tuxedo Suite 26:
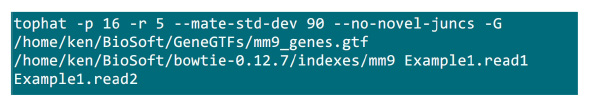
- Ordnen Sie die Paired-End-RNA-Seq liest zur Maus Referenzgenom (UCSC Version MM9, erhalten aus http://cufflinks.cbcb.umd.edu/igenomes.html ) mit Tophat-Software (Version 1.3.3) 27, die verwendet die Bowtie lesen Mapper (Version 0.12.7) 28. Tophat mit der Option "-no-Roman-juncs", um die Schätzgenauigkeit der Expressionsniveau verbessern geliefert.
- Setzen Sie die .fastq Dateien in einem Ordner, in dem die Mapping-Prozess umgesetzt werden. Angenommen, es gibt zwei .fastq Dateien (umbenennen Example1.read1, Example1.read2) für eine Paired-End-Sequenzierung Probe, verwenden Sie den Befehl in Abbildung S2, um die Zuordnung (passen Sie die Parameter entsprechend der Systemeinstellung) zu tun.Der Parameter "-p" liefert die CPU-Nummer, die verwendet werden. Die "-r" und "-mate-STD-dev" Parameter können aus der Bibliothek QC erhalten oder aus einer Teilmenge ausgerichtet gefolgert liest (Abbildung S2) werden.
- Montieren Sie den abgebildeten liest in RNA-Transkripte mit der Manschettenknöpfe-Software (Version 1.3.0) 29. Führen Manschettenknöpfe mit der Anmerkungsdatei von bekannten Genen (gleiche .GTF Datei Tophat verwendet) und .bam Datei Tophat produziert.
- Nach dem Ausführen Tophat abgeschlossen, im selben Ordner, verwenden Sie den Befehl in Abbildung S3A um Manschettenknöpfe laufen Transkriptom und Schätzung Transkript Expressionsniveau zu konstruieren. Die 'mm9_repeatMasker.gtf' und Genomsequenz-Dateien im Ordner "GenomeSeqMM9 'kann von UCSC Genome Browser erreicht werden.
- Die resultierenden genes.expr und transcripts.expr Dateien enthalten den Ausdruckswert der Gene und Transkripte (Isoformen). Kopieren und Einfügender Inhalt der Datei in eine Excel-Datei mit Tabellenkalkulationsprogramm (Abbildung S3B) zu manipulieren.
- Verwenden Sie den Befehl in Abbildung S3C die resultierende Datei 'transcripts.gtf' zu dem Aktenzeichen "mm9_genes.gtf 'um neue Transkripte identifizieren zu vergleichen.
- Die resultierende Datei enthält .tmap dem Vergleichsergebnis. Kopieren und fügen Sie den Inhalt der Datei in eine Excel-Datei und mit Tabellenkalkulationsprogramm zu bearbeiten. Transkripte mit Klassencode 'u' können als "neue" gegenüber dem Referenz .GTF Datei bereitgestellt (Abbildung S3D) berücksichtigt werden.
HINWEIS: Für nachgeschaltete Analyse Bequemlichkeit, setzen Sie die FPKM Werte bis 0,1, wenn die Werte unter 0,1.
HINWEIS: Schritt 3.2.3 - 3.2.6 ist optional für diejenigen, die die Genauigkeit des Ausdrucks Schätzung neuer Transkripte 'verbessern möchten. Dadurch wird eine wesentlich längere Zeit in Anspruch nehmen, weil Kartierung und Transkriptom Konstruktion müssen run mehr als einmal.
- Führen Tophat mit Default-Parameter und führen Sie Manschettenknöpfe erzeugt .GTF Datei mit dem Befehl in Abbildung S3E.
- Vergleichen Sie die entstandene .GTF Datei in das Referenzgenom .GTF Datei mit dem Befehl in Abbildung S3F.
- Analysieren Sie die in Folge .tmap Datei wie in Schritt 3.2.2.4 beschrieben. Kopieren und fügen Sie den Inhalt der Datei in eine Excel-Datei und mit Tabellenkalkulationsprogramm zu bearbeiten. Transkripte mit Klassencode 'u' kann als betrachtet werden "Roman" im Vergleich zum Referenz .GTF Datei zur Verfügung gestellt.
- Nach dem Schritt 3.2.5 gibt es eine .combined.gtf Datei in dem Ordner, der als Referenz .GTF Datei verwendet werden kann. Eine zweite Auflage von Tophat und Manschettenknöpfe können wie in Schritt 3.2.1 und 3.2.2 beschrieben, um eine genauere FPKM Abschätzung neuer Transkripte zu erhalten durchgeführt werden.
- Ordnen Sie die Paired-End-RNA-Seq liest zur Maus Referenzgenom (UCSC Version MM9, erhalten aus http://cufflinks.cbcb.umd.edu/igenomes.html ) mit Tophat-Software (Version 1.3.3) 27, die verwendet die Bowtie lesen Mapper (Version 0.12.7) 28. Tophat mit der Option "-no-Roman-juncs", um die Schätzgenauigkeit der Expressionsniveau verbessern geliefert.
- Detect differentially exprimierten Gene mit DESeq Paket 30.
- Der Eingang des DESeq ist eine rohe Lese zählt Tisch. Um eine solche Tabelle zu erhalten, verwenden Sie die htseq-count Skript mit dem HTSeq Python-Paket, das aus HTSeq Website heruntergeladen werden kann verteilt ( http://www-huber.embl.de/users/anders/HTSeq/doc/count.html ) .
- Stellen Sie sicher, dass samtools, Python und htseq-count programsare im System installiert. Erhalten rohen Lesezählung Zahlen von tophat Ausgabe mit dem Befehl in Abbildung S4A.
- Bereiten 'Raw_Count_Table.txt', 'ExperimentDesign.txt' Dateien mit Excel. Kopieren und speichern Sie den Inhalt in TXT-Format für die DESeq R-Paket (Abbildung S4B).
- Installieren R Programm im System. Im Terminal, zeigten Typ 'R' und drücken ENTER.A Bildschirmmeldung wird appearas in Abbildung S4C.
- Lesen Sie 'Raw_Count_Table.txt ',' ExperimentDesign.txt 'in R mit dem Befehl in Abbildung S4D.
- Legen DESeq Paket mit dem Befehl in Abbildung S4E.
- Factorize Bedingungen in R (Abbildung S4F).
- Verwenden Sie den Befehl in Abbildung S4G negative Binominal Test auf dem normalisierten Zähltabelle laufen.
- Verwenden Sie den Befehl in Abbildung S4H ausgegeben signifikante Differenz exprimierten Genen in einer CSV-Datei.
- Der Eingang des DESeq ist eine rohe Lese zählt Tisch. Um eine solche Tabelle zu erhalten, verwenden Sie die htseq-count Skript mit dem HTSeq Python-Paket, das aus HTSeq Website heruntergeladen werden kann verteilt ( http://www-huber.embl.de/users/anders/HTSeq/doc/count.html ) .
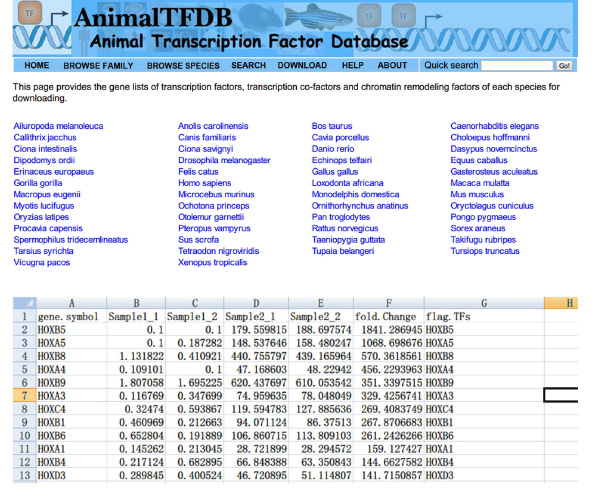
- Lookup Transkriptionsfaktoren "(TFS) FPKM Werte über Proben mit Hilfe von Excel. Intersect DE Gen Tisch und TFs Tisch. Gene gehören beiden Tabelle sind differentiell exprimiert Transkriptionsfaktoren.
- Besuchen Sie die Website http://www.bioguo.org/AnimalTFDB/download.php und laden Sie die Transkriptionsfaktoren. Dann Lookup der DE-Transkriptionsfaktoren in der Excel (< strong> Abbildung S5).
- Erzeugen .bigwig Datei für UCSC Genome Browser Visualisierung.
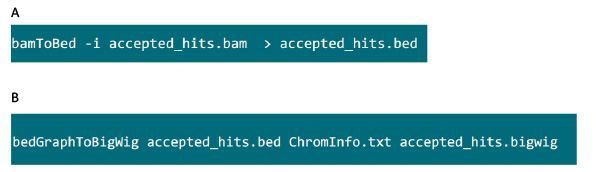
- Download 'bedtools' Softwarepaket von der Website https://github.com/arq5x/bedtools2 und installieren Sie die Software im System 31. Laden Sie die UCSC Tools 'bedGraphToBigWig' von der Website http://hgdownload.cse.ucsc.edu/admin/exe/ und installieren Sie die Software im System.
- In dem Ordner mit dem .bam Datei, verwenden Sie den Befehl in Abbildung S6A zu .bam Datei tophat in .bed Datei generiert konvertieren.
- Nachdem die .bed Datei erzeugt wird, verwenden Sie den Befehl in Abbildung S6B zu .bigwig Datei zu generieren. Die Datei 'ChromInfo.txt' kann von folgender URL erhältlich:Arget = "_blank"> http://hgdownload.cse.ucsc.edu/goldenPath/mm9/database/chromInfo.txt.gz.
- Beachten Sie eine benutzerdefinierte Spur auf UCSC Genome Browser. Siehe die Website http://genome.ucsc.edu/goldenPath/help/customTrack.html auf, wie eine benutzerdefinierte Spur mit UCSC Genom-Browser anzuzeigen.

Abbildung S1: Konvertieren .bcl Datei Datei auf .fastq Verwendung casava Software.

Abbildung S2: Mapping liest Genom Referenz mit Tophat.
Abbildung S3: Nachweis von neuer Transkripte und Expressionsniveau Schätzung.

Abbildung S4: Aufruf Differential exprimierte Gen mit DESeq Paket.

Abbildung S5: Bezeichnung des differentiell exprimierten Transkriptionsfaktoren.

Abbildung S6: Konvertieren Mapping-Ergebnis für die Datenvisualisierung.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Um differenziell exprimierte Gene in Lin-CD34 + und Lin-CD34- EML Zellen zu analysieren, haben wir RNA-Seq-Technologie. Abbildung 1 zeigt den Workflow der Verfahren. Nach der Isolierung von Lineage-negativen Zellen durch magnetische Zellsortierung, trennten wir Lin-SCA + CD34 + und Lin-SCA-CD34- Zellen mittels FACS Aria. Lin angereicherte EML-Zellen wurden mit anti-CD34, anti-Sca1 und Abstammung cocktail Antikörpern gefärbt. Nur Lin- Zellen wurden für die Analyse von Sca1 und CD34 Expression gated. Zwei Populationen (SCA + CD34 + und CD34- SCA-EML-Zellen) durch FACS-Analyse (Figur 2) 6 beobachtet werden.
Nach Zellseparation, extrahiert wir Gesamt-RNA aus CD34 + und CD34- Zellen jeweils und analysiert die Qualität der RNA. Die Genauigkeit der RNA-Seq Daten beruht vorwiegend auf die Qualität der RNA-Seq-Bibliothek und die Qualität der Gesamt-RNA ist entscheidend für die Herstellung eines qualitativ hochwertigen Bibliothek. Hochwertige RNA-Probe sollte einen OD 260/280 Wert zwischen 1 haben.8 und 2,0. Zusätzlich zur Verwendung des Spektrophotometers wurde RNA Qualität weiter mit größerer Genauigkeit durch Bioanalyzer beurteilt. 3 zeigt ein Ergebnis einer hohen Qualität RNA-Probe mit dem RIN gleich 9,4. Nur qualitativ hochwertige RNA-Probe mit RIN-Wert größer als 9 wurde für die mRNA-Extraktion und anschließende Bibliothek Bauverfahren eingesetzt.
Ribosomale RNA ist die häufigste Art von RNA in der Zelle. Derzeit sind zwei Hauptstrategien, Erschöpfung der rRNA oder positiv Auswahl polyadenyliert mRNA (Poly-A-mRNA), sind für die Anreicherung von Ziel-RNA vor Bibliothekskonstruktion verwendet. Nicht polyadenylierte RNA-Spezies sind bei der Auswahl der Poly-A-mRNA verloren. Im Gegensatz dazu konnte rRNA Depletion Methoden wie RiboMinus nicht polyadenylierten RNA-Spezies zu bewahren. Das Ziel unserer Studie ist es für differentiell exprimierte kodierenden Gene in beiden Zelltypen, so verwendet man die Poly-A-mRNA Selektionsverfahren zur Anreicherung der Ziel-RNAs, bevor Bibliothek constru aussehenction. Wenn Bibliothekskonstruktion beendet war, wurde die Größe der DNA-Fragmente in der Bibliothek vor Sequenzierung unter Verwendung Bioanalyzer geprüft. Figur 4 zeigt eine gute Qualität Bibliothek mit den Fragmentgröße Peaks bei etwa 300 bp.
In dem nachfolgenden Schritt wurde die Bibliothek auf Hochdurchsatz-Sequenzierung unterzogen. Im Prinzip wird mehr gelesen Länge hilfreich für Lese Abbildung. Es kann die Wahrscheinlichkeit verringern, dass der Lesevorgang auf mehrere Standorte wegen Ähnlichkeit zwischen doppelten Gene oder Gensequenzen Familienmitglieder abgebildet. Da die Paar-End-Sequenzierung Sequenzen von beiden Enden der Fragmente, sollte die gewählte Leselänge weniger als die Hälfte der durchschnittlichen Länge von Fragmenten. Wenn das Hauptziel des Experiments ist es, das Expressionsniveau statt Konstruieren Transkript Struktur messen, single-Ende gelesen (75 oder 100 bp) können die Kosten, ohne dass zu viel Information zu reduzieren. Paired-End-Sequenzierung ist nützlicher für Transkript Strukturaufbau und kürzereLeselänge verwendet werden, um Kosten zu reduzieren. Gewiss, wenn ausreichend Mittel zur Verfügung stehen, wird bevorzugt, mehr Leselänge.
Für Differenzexpressionsanalyse, gibt es viele andere Möglichkeit, als DESeq Algorithmen. Es gibt auch eine in Manschettenknöpfe Paket cuffdiff 32 Namen enthalten. DESeq ist eine der am weitesten verbreiteten Zahl basierend DE-Gen-Analyse-Algorithmen. Negative Binomialverteilung - DESeq Methode basiert auf einem gut charakterisierten Statistik-Modell. Nach unserer Erfahrung ist DESeq stabiler zu vergleichen, um cuffdiff. Frühe Versionen von cuffdiff geben oft deutlich unterschiedliche Anzahlen von DE-Gene. Daher verwendeten wir DESeq für DE Analyse hier.
Da Transkriptionsfaktoren sind entscheidend für das Schicksal der Zelle Bestimmung, auf der deutlich differenziell exprimierte Transkriptionsfaktoren haben wir uns 33. TFS verändert> 1.5 Falte zwischen Lin-CD34 + und Lin-CD34- wurden gefunden und werden auf der Heatmap dargestellt (Figure 5) 2. Insbesondere ist die relative Expressionsniveau TCF7 in Lin-CD34 + Zellen mehr als 100-fach höher als die in Lin-CD34- Zellen. So wurde TCF7 zur weiteren ChIP-Sequenzierung (Chromatin Immunopräzipitation und Sequenzierung) Analyse und Funktionstest gewählt, um TCF7 Schen Funktion in der Regulation der EML Zellselbsterneuerung und Differenzierung 2 bestätigen.

Fig. 1: Arbeitsablauf der Verfahren Lin-CD34 + und CD34- Lin-Zellen durch magnetische Zellseparation System und Fluoreszenz-aktivierten Zellsortierverfahren getrennt. Gesamt-RNA wurde extrahiert, gefolgt von mRNA Reinigung und die Konstruktion der Bibliothek. Nach Analyse der Qualität von Bibliotheken wurden die Proben auf Hochdurchsatz-Sequenzierung unterzogen. Die Daten wurden analysiert und differentiell exprimierte Transkriptionsfaktoren identifiziert wurden.

Abbildung 2: Trennung von Lin-CD34 + und Lin-CD34- EML Zellen 6 Lin- EML Zellen wurden durch magnetische Zellsortierung angereichert.. Lin- Zellen wurden mit anti-CD34, anti-Sca1 und Abstammung Mischung Antikörpern gefärbt. Lin- Zellen wurden für die Expression von CD34 und Sca1 gated. Lin-CD34 + SCA + und Lin-CD34-SCADA EML Zellpopulationen wurden sortiert.

Fig. 3: Eine repräsentative hochwertiger RNA-Probe Die Qualität der Gesamt-RNA wurde durch Bioanalyzer beurteilt. Die RNA Integrity Number ist 9,4 (FU, Fluorescence Units).

Abbildung 4:. Fragmente Größenbereich von Paired-End-Bibliothek Die DNA-Größenverteilung der Bibliothek wurde unter Verwendung Bioanalyzer analysiert. Die meisten Fragmente im Größenbereich von 250-500 bp.

Fig. 5: Differentiell exprimierte Transkriptionsfaktoren (> 1,5-fach) von Lin-CD34 + Zellen und Lin-CD34- Zellen 2 für jeden Zelltyp, zwei unabhängige Experimente durchgeführt. Up-regulierte Gene sind angegeben als rote Farbe und down-regulierten Gene werden als grüne Farbe angezeigt.
Tabelle 1: Puffer und Zellkulturmedien.
| Software | Verwendung | Referenz | |||
| Bowtie 1.2.7 | Wird von Tophat zur Kartierung | [28] | |||
| Tophat 1.3.3 | Mapping liest Zurück zur Referenzgenom | [27] | |||
| Manschettenknöpfe 1.3.0 | Transkripte Konstruktion und Expression-Schätz | [29] | |||
| DESeq 1.16.0 | Differentielle Expression Analyse | [30] | Bedtools 2.18 | .bam Datei zu konvertieren in .bed Datei | [31] |
| bedGraphToBigWig | .bed Datei zu Datei .bigwig umrechnen | http://genome.ucsc.edu/ |
Tabelle 2: Liste der Software für die Datenanalyse.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Mammalian Transkriptom ist sehr komplex 34-38. RNA-Seq-Technologie spielt eine immer wichtigere Rolle in den Untersuchungen der Transkriptomanalyse, neuer Transkripte Erkennung und single nucleotide Variation Entdeckung etc. Es hat viele Vorteile gegenüber anderen Methoden zur Genexpressionsanalyse. Wie in der Einleitung erwähnt, überwindet sie die Hybridisierung Artefakte der Mikroarray und kann verwendet werden, um neue Transkripte de novo zu identifizieren. Eine Einschränkung der RNA-Sequenzierung ist relativ kurzen Leselänge im Vergleich zu Sanger-Sequenzierung. Doch mit der raschen Verbesserung der Sequenzierungstechnik, Leselänge nimmt ständig zu. In diesem Beitrag stellen wir detaillierte Verfahren zur Verwendung dieser Technologie, um potenzielle Schlüsselregulatoren in der Maus EML Zellselbsterneuerung und Differenzierung zu identifizieren.
Der erste Schlüsselschritt für dieses Protokoll ist EML Zellkultur. Obwohl EML eine hämatopoetische Vorläuferzell-Linie und es kannin großer Menge mit SCF propagiert. Die Kultivierung Zustand EML Zellen erfordert mehr Aufmerksamkeit als die üblichen immortalisierten Zelllinien. Die Zellen sollten eingespeist und bei regelmäßig mit sanften Betrieb agiert werden; andernfalls werden die Zellen konnten in ihren Eigenschaften zur Selbsterneuerung und Differenzierung verändern und Zelltod. Als erster Schritt nach dem Sammeln von genügend Zellen, isolierten wir Linie negativen Zellen unter Verwendung eines magnetischen Zellsortierung System. Dann abgetrennt wir CD34 + und CD34- Zellen mittels fluoreszenzaktivierter Zellsortierung. Die EML-Zellen werden in der Regel vor der Verwendung für die RNA-Extraktion und die Zahl der CD34 + und CD34- Zellen sollte ähnlich nach der Trennung sein agiert weniger als 10 Generationen. Wenn die beiden Populationen variieren stark in der Zellzahl, ist es ratsam, die Kultur zu verwerfen und neu zu tauen anderes Rohr Zell Lager für Kultur.
Nach der Trennung von CD34 + und CD34- Zell wurde Gesamt-RNA-Extraktion durchgeführt wird, ein weiterer wichtiger Schritt für diese study. Hochwertige RNA ist die Basis für den Bau eines hochwertigen Bibliothek, die die Genauigkeit der Sequenzierungsdaten verspricht. In diesem kritischen Schritt sollte jeder Kontakt mit RNase zu vermeiden. Alle Reagenzien sollten frei RNase werden. Es ist wichtig, Handschuhe jederzeit zu tragen beim Umgang mit RNA. Hochwertige RNA-Probe hat eine OD 260/280 Wert zwischen 1,8 und 2,0. Beim Sammeln der wässrigen Phase, die RNA, seien Sie vorsichtig, keine organischen Phase mit der RNA-Probe zu tragen. Alle restlichen organischen Lösungsmitteln, wie Phenol oder Chloroform in RNA würde in einem OD260 / 280-Wert niedriger als 1,65 führen. Wenn der OD260 / 280-Wert niedriger als 1,65 ist, auszufällen RNA erneut mit Ethanol. Nach Waschen mit 75% Ethanol, nicht übertrocknen RNA-Pellet. Trocknen RNA-Pellet wird komplett die Löslichkeit der RNA beeinflussen und zu geringe Ausbeute an RNA.
Der nächste wichtige Schritt ist dieses Protokoll Bibliothek Vorbereitung. Nach Gesamt-RNA-Extraktion, eine Stufe der Verwendung DNase zum Entfernen kontaminierter DNA is sehr zu empfehlen, da DNA-Kontamination könnte in falsche Schätzung der Menge an Gesamt-RNA verwendet führen. Es wird empfohlen, das stromabwärts Verfahren unmittelbar nach der RNA-Isolierung durchzuführen, da bei Langzeitlagerung und Frost-Tau-Verfahren wird RNA zu einem gewissen Grad verschlechtert. Wenn die nachfolgenden Schritte nach der RNA-Isolierung nicht sofort durchgeführt werden, speichern Sie die RNA in -80 ° C. Bevor die Gesamt-RNA wird für mRNA Reinigung und cDNA-Synthese verwendet, sollte die Qualität immer überprüft werden. Nur qualitativ hochwertige RNA kann für Bibliotheks Zubereitung verwendet werden. Mit geringer Qualität oder degradierten RNA könnte zu Überrepräsentation von 3'-Enden führen. Bevor Sequenzierung wurde Bibliotheksqualität beurteilt, um eine maximale Effizienz zu gewährleisten Sequenzierung.
In der Datenanalyse Teil, nach der Durchführung einer Auflage von Manschettenknöpfe ohne Referenz Transkriptom, kombinierten wir die neuen Transkripte mit bekannten Transkripte um einen Verweis .GTF Datei und starten Tophat und Manschettenknöpfe zum zweiten Mal zu bilden.Dieses Zwei-Run Vorgehen empfohlen, da dies eine genauere FPKM Schätzung als nur einmal ausgeführt wird. Nachdem die Datenanalyse wurden die differentiell exprimierten Gene identifiziert. Downstream-Experimente können durchgeführt werden, um die Funktion von Genen in vitro und in vivo belegen. In unserer früheren Veröffentlichung 2, wählten wir die signifikant differentiell exprimiert Transkriptionsfaktoren und identifiziert das Genom Bindungsstelle dieser Faktoren durch Ausführen Chromatinimmunpräzipitation und Sequenzierung (ChIP-Seq). Zusätzlich verwendeten wir shRNA Zuschlags Assay, um die funktionelle Wirkung TCF7 testen. Wir fanden, dass in TCF7 Knockdown-Zellen, waren hochreguliert Gene die Gene in CD34- Zellen hoch angereichertes, während nach unten reguliert Gene erwiesen sich deutlich in CD34 + -Zellen angereichert werden. Daher das Genexpressionsprofil von TCF7 Zuschlags Zellen verschoben hin zu einer teilweise differenzierte CD34- state.Overall mit EML Zelle als Modellsystemgepaart mit RNA-Sequencing Technologie und funktionelle Assays, wir identifiziert und bestätigt TCF7 als wichtiger Regulator der EML Zellselbsterneuerung und Differenzierung.
Subscription Required. Please recommend JoVE to your librarian.
Materials
| Name | Company | Catalog Number | Comments |
| Antibiotic-Antimycotic | Invitrogen | 15240-062 | BHK cell culture |
| Anti-Mouse CD34 FITC | eBioscience | 11-0341-81 | FACS sorting |
| Anti-Mouse Ly-6A/E (Sca-1) PE | eBioscience | 12-5981-81 | FACS sorting |
| APC Mouse Lineage Antibody Cocktail | BD Biosciences | 558074 | FACS sorting |
| BD FACSAria Cell Sorter | BD Biosciences | Special offer sysmtem | FACS sorting |
| Corning™ Cell Culture Treated Flasks 75 cm2 | Corning incorporated | 430641 | Cell culture |
| Corning™ Cell Culture Treated Flasks 25 cm2 | Corning incorporated | 430639 | Cell culture |
| Deoxyribonuclease I, Amplification Grade | Invitrogen | 18068-015 | Library preparation |
| DMEM | Invitrogen | 11965-092 | BHK cell culture |
| DPBS | Gibco | 14190 | Cell culture |
| HI FBS | Invitrogen | 16140071 | BHK cell culture |
| Horse Serum | Invitrogen | 16050-122 | EML cell culture |
| IMDM | HyClone | SH30228.02 | EML cell culture |
| L-Glutamine | Invitrogen | 25030-081 | Cell culture |
| Lineage Cell Depletion Kit, mouse | Miltenyi Biotec | 130-090-858 | Isolation of lineage negative cells |
| NanoVue Plus spectrophotometer | GE Healthcare | 28-9569-62 | Quality control |
| Thermo Scientific™ Napco™ 8000 Water-Jacketed CO2 Incubators | Thermo Scientific | 15-497-002 | Cell culture |
| Penicillin-Streptomycin | Invitrogen | 15140-122 | EML cell culture |
| TRIzol® Reagent | Invitrogen | 15596-018 | RNA exraction |
| TruSeq™ RNA Sample Prep Kit v2 -Set B (48 rxn) | Illumina | RS-122-2002 | Library preparation |
| 2100 Electrophoresis Bioanalyzer Instrument | Agilent | G2939AA | Quality control |
| 0.25% Trypsin-EDTA | Gibco | 25200 | Cell culture |
| 0.45 µm Syringe Filters | Nalgene | 190-2545 | Cell culture |
References
- Chambers, S. M., Goodell, M. A. Hematopoietic stem cell aging: wrinkles in stem cell potential. Stem Cell Rev. 3, 201-211 (2007).
- Wu, J. Q., et al. Tcf7 is an important regulator of the switch of self-renewal and differentiation in a multipotential hematopoietic cell line. PLoS genetics. 8, (2012).
- Ye, Z. J., et al. Complex interactions in EML cell stimulation by stem cell factor and IL-3. Proceedings of the National Academy of Sciences of the United States of America. 108, 4882-4887 (2011).
- Tsai, S., Bartelmez, S., Sitnicka, E., Collins, S. Lymphohematopoietic progenitors immortalized by a retroviral vector harboring a dominant-negative retinoic acid receptor can recapitulate lymphoid, myeloid, and erythroid development. Genes Dev. 8, 2831-2841 (1994).
- Weiler, S. R., et al. D3: a gene induced during myeloid cell differentiation of Linlo c-Kit+ Sca-1(+) progenitor cells. Blood. 93, 527-536 (1999).
- Ye, Z. J., Kluger, Y., Lian, Z., Weissman, S. M. Two types of precursor cells in a multipotential hematopoietic cell line. Proc Natl Acad Sci U S A. 102, 18461-18466 (2005).
- Wang, Z., Gerstein, M., Snyder, M. RNA-Seq: a revolutionary tool for transcriptomics. Nature reviews. Genetics. 10, 57-63 (2009).
- Chu, Y., Corey, D. R. RNA sequencing: platform selection, experimental design, and data interpretation. Nucleic acid therapeutics. 22, 271-274 (2012).
- Hornett, E. A., Wheat, C. W. Quantitative RNA-Seq analysis in non-model species: assessing transcriptome assemblies as a scaffold and the utility of evolutionary divergent genomic reference species. BMC genomics. 13, 361 (2012).
- Eswaran, J., et al. RNA sequencing of cancer reveals novel splicing alterations. Scientific reports. 3, 1689 (2013).
- Wang, E. T., et al. Alternative isoform regulation in human tissue transcriptomes. Nature. 456, 470-476 (2008).
- Wu, J. Q., et al. Dynamic transcriptomes during neural differentiation of human embryonic stem cells revealed by short, long, and paired-end sequencing. Proceedings of the National Academy of Sciences of the United States of America. 107, 5254-5259 (2010).
- Loraine, A. E., McCormick, S., Estrada, A., Patel, K., Qin, P. RNA-seq of Arabidopsis pollen uncovers novel transcription and alternative splicing. Plant physiology. 162, 1092-1109 (2013).
- Edgren, H., et al. Identification of fusion genes in breast cancer by paired-end RNA-sequencing. Genome biology. 12, 6 (2011).
- Ilott, N. E., Ponting, C. P. Predicting long non-coding RNAs using RNA sequencing. Methods. 63, 50-59 (2013).
- Sun, L., et al. Prediction of novel long non-coding RNAs based on RNA-Seq data of mouse Klf1 knockout study. BMC bioinformatics. 13, 331 (2012).
- Luo, S. MicroRNA expression analysis using the Illumina microRNA-Seq Platform. Methods in molecular biology. 822, 183-188 (2012).
- Bolduc, F., Hoareau, C., St-Pierre, P., Perreault, J. P. In-depth sequencing of the siRNAs associated with peach latent mosaic viroid infection. BMC molecular biology. 11, 16 (2010).
- Chepelev, I., Wei, G., Tang, Q., Zhao, K. Detection of single nucleotide variations in expressed exons of the human genome using RNA-Seq. Nucleic acids research. 37, 106 (2009).
- Djari, A., et al. Gene-based single nucleotide polymorphism discovery in bovine muscle using next-generation transcriptomic sequencing. BMC genomics. 14, 307 (2013).
- Murphy, D. Gene expression studies using microarrays: principles, problems, and prospects. Advances in physiology education. 26, 256-270 (2002).
- Chen, K., et al. RNA-seq characterization of spinal cord injury transcriptome in acute/subacute phases: a resource for understanding the pathology at the systems level. PLoS one. 8, 72567 (2013).
- Marioni, J. C., Mason, C. E., Mane, S. M., Stephens, M., Gilad, Y. RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome research. 18, 1509-1517 (2008).
- Ramskold, D., Kavak, E., Sandberg, R. How to analyze gene expression using RNA-sequencing data. Methods in molecular biology. 802, 259-274 (2012).
- Glenn, T. C. Field guide to next-generation DNA sequencers. Mol Ecol Resour. 11, 759-769 (2011).
- Trapnell, C., et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nature protocols. 7, 562-578 (2012).
- Trapnell, C., Pachter, L., Salzberg, S. L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 25, 1105-1111 (2009).
- Langmead, B., Trapnell, C., Pop, M., Salzberg, S. L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome biology. 10, 25 (2009).
- Trapnell, C., et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature. 28, 511-515 (2010).
- Anders, S., Huber, W. Differential expression analysis for sequence count data. Genome biology. 11, 106 (2010).
- Quinlan, A. R., Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 26, 841-842 (2010).
- Cheranova, D., et al. RNA-seq analysis of transcriptomes in thrombin-treated and control human pulmonary microvascular endothelial cells. J Vis Exp. , (2013).
- Zhang, H. M., et al. AnimalTFDB: a comprehensive animal transcription factor database. Nucleic acids research. 40, 144-149 (2012).
- Wu, J. Q., et al. Systematic analysis of transcribed loci in ENCODE regions using RACE sequencing reveals extensive transcription in the human genome. Genome Biol. 9, 3 (2008).
- Wu, J. Q., et al. Large-scale RT-PCR recovery of full-length cDNA clones. Biotechniques. 36, 690-696 (2004).
- Wu, J. Q., Shteynberg, D., Arumugam, M., Gibbs, R. A., Brent, M. R. Identification of rat genes by TWINSCAN gene prediction, RT-PCR, and direct sequencing. Genome Res. 14, 665-671 (2004).
- Dewey, C., et al. Accurate identification of novel human genes through simultaneous gene prediction in human, mouse, and rat. Genome Res. 14, 661-664 (2004).
- Wu, J. Characterize Mammalian Transcriptome Complexity. , LAP Lambert Academic Publishing. (2011).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}