

Summary
RNA-sekventering og bioinformatik analyser blev anvendt til at identificere betydeligt, differentielt udtrykte transkriptionsfaktorer i Lin-CD34 + Lin-CD34 subpopulationer af mus EMLcells. Disse transkriptionsfaktorer kan spille en vigtig rolle i fastsættelsen af kontakten mellem selvfornyende Lin-CD34 + og delvist differentierede Lin-CD34-celler.
Abstract
Hæmatopoietiske stamceller (HSC'er) anvendes klinisk til transplantation behandling at genopbygge en patients hæmatopoietiske system i mange sygdomme såsom leukæmi og lymfom. Belyse de mekanismer, der styrer HSC'er selvfornyelse og differentiering er vigtig for anvendelsen af HSC'er for forskning og kliniske anvendelser. Dog er det muligt at opnå store mængder af HSC'er på grund af deres manglende evne til at proliferere in vitro. At overvinde denne forhindring, vi anvendte en muse knoglemarvsafledte cellelinie EML (erythroid, myeloid og lymfatisk) cellelinie, som en model for denne undersøgelse.
RNA-sekventering (RNA-Seq) er i stigende grad blevet anvendt til at erstatte microarray for genekspressionsstudier. Vi rapporterer her en detaljeret metode til at bruge RNA-Seq-teknologi til at undersøge de potentielle nøglefaktorer i reguleringen af EML celle selvfornyelse og differentiering. Oplysningerne i dette papir protokol er opdelt i tre dele. Det første part forklarer, hvordan kultur EML celler og separat Lin-CD34 + og Lin-CD34-celler. Den anden del af protokollen giver detaljerede procedurer for total RNA forberedelse og den efterfølgende bibliotek konstruktion for high-throughput sekventering. Den sidste del beskriver fremgangsmåden for RNA-Seq dataanalyse og forklarer, hvordan man bruger data til at identificere differentielt udtrykte transkriptionsfaktorer mellem Lin-CD34 + og Lin-CD34-celler. De mest markant differentielt udtrykte transkriptionsfaktorer blev identificeret til at være de potentielle vigtige regulatorer styrer EML celle selvfornyelse og differentiering. I diskussionen del af dette dokument, vi fremhæve de vigtigste skridt for en vellykket gennemførelse af dette eksperiment.
Sammenfattende er dette papir tilbyder en metode til at bruge RNA-Seq teknologi til at identificere potentielle regulatorer af selvfornyelse og differentiering i EML celler. De vigtigste faktorer, der er udsat for downstream funktionel analyse in vitro og in vivo.
Introduction
Hæmatopoietiske stamceller er sjældne blodceller, som hovedsagelig bor i den voksne knoglemarv niche. De er ansvarlige for produktionen af nødvendige for at genopbygge blod celler og immunforsvar 1. Som en slags stamceller, HSC'er er i stand til både selvfornyelse og differentiering. Belyse mekanismer, der styrer skæbne afgørelse af HSC'er, mod enten selv-fornyelse eller differentiering, vil tilbyde værdifuld vejledning om manipulation af HSC'er for blodsygdom forsker og klinisk brug 2. Et problem, som forskerne står over for, er, at HSC'er kan fastholdes og udbygges in vitro i meget begrænset omfang; størstedelen af deres afkom er delvist differentieret i kultur 2.
Med henblik på at identificere de vigtigste regulatorer, der styrer de processer af selv-fornyelse og differentiering ved en genom-plan, vi brugte en mus primitive hæmatopoietiske cellelinie EML som modelsystem. Ther cellelinie blev afledt fra murin knoglemarv 3,4. Når de fodres med forskellige vækstfaktorer, kan EML-celler differentierer i erythroid, myeloid og lymfoide celler in vitro 5. Vigtigere, kan denne cellelinie opformeres i store mængder i dyrkningsmedium indeholdende stamcelle faktor (SCF) og stadig bevarer deres multipotentiality. EML-celler kan adskilles i subpopulationer af selvfornyende Lin-SCA + CD34 + og delvist differentierede Lin-SCA-CD34 celler baseret på overflademarkører CD34 og SCA 6. Svarende til kortsigtede HSC'er, SCA + CD34 + celler er i stand til selv-fornyelse. Ved behandling med SCF, Lin-SCA + CD34 + celler kan hurtigt regenerere en blandet population af Lin-SCA + CD34 + Lin-SCA-CD34-celler og fortsætte med at formere 6. De to populationer er ens i morfologi og har tilsvarende niveauer af c-kit-mRNA og protein 6. Lin-SCA-CD34 celler er i stand til at udbrede i medier indeholdende IL-3 i stedet for SCF3. Unveiling de centrale tilsynsmyndigheder i EML celleskæbnen beslutning vil tilbyde en bedre forståelse af cellulære og molekylære mekanismer i begyndelsen udviklingsmæssige overgang under hæmatopoiese.
For at undersøge de underliggende molekylære forskelle mellem selvfornyende Lin-SCA + CD34 + og delvist differentierede Lin-SCA-CD34-celler anvendte vi RNA-Seq at identificere differentielt udtrykte gener. I særdeleshed har vi fokus på transkriptionsfaktorer, som transkriptionsfaktorer er afgørende for celle skæbne. RNA-Seq er en nyudviklet metode, der udnytter mulighederne i næste generation sekventering (NGS) teknologier til at profilere og kvantificere RNA'er transskriberet fra genom 7,8. Kort fortalt, total RNA er poly-A-udvalgt og fragmenteret som den første template.The RNA-template derefter omdannes til cDNA under anvendelse af revers transkriptase. For at kortlægge fuld længde RNA-transkripter ved anvendelse af intakt, ikke-nedbrudt RNA til konstruktion af cDNA-bibliotek er vigtig. For purudgøre af sekventering specifikke adaptorsekvenser tilsat til begge ender af cDNA. Så i de fleste tilfælde, er cDNA-molekyler amplificeret ved PCR og sekventeret i en high-throughput måde.
Efter sekventering, læser den resulterende kan justeres til en reference genom og en transkriptom database. Antallet af læser, at kortet til reference-genet tælles og denne information kan anvendes til at estimere genekspression niveau. Den læser kan også samles de novo uden henvisning genom muliggør studiet af transcriptomes i ikke-modelorganismer 9. RNA-seq teknologi er også blevet anvendt til at detektere splejsningsisoformer 10-12, nye transkripter 13 og genfusioner 14. Ud over påvisning af protein-kodende gener, kan RNA-Seq også anvendes til at påvise hidtil ukendte og analysere transkription niveau af ikke-kodende RNA, såsom lang ikke-kodende RNA 15,16, microRNA 17 siRNA etc. 18. På grund af than nøjagtighed af denne fremgangsmåde, er det blevet anvendt til påvisning af enkelt nucleotidvariationer 19,20.
Før fremkomsten af RNA-Seq teknologi, microarray var den vigtigste metode til analyse af genekspression profil. Pre-designede prober syntetiseres og efterfølgende fastgøres til en fast overflade for at danne en microarray slide 21. mRNA ekstraheres og omdannes til cDNA. Under revers transkription proces, fluorescensmærkede nukleotider inkorporeret i cDNA'et, og cDNA kan hybridiseres onto microarray slides. Intensiteten af signalet opsamlet fra et bestemt sted, afhænger af mængden af cDNA-binding til den specifikke probe på dette sted 21. Sammenlignet med RNA-Seq teknologi, microarray har flere begrænsninger. Først microarray bygger på allerede eksisterende viden om gen-anmærkning, mens RNA-Seq teknologi er i stand til at opdage hidtil ukendte udskrifter ved en relativ høj baggrund niveau, hvilket begrænser dets brug, når GEne udtryk er lavt. Desuden RNA-Seq teknologi har langt højere dynamikområde detektion (8000 gange) 7, mens grund baggrund og mætning af signaler, er begrænset for både højt og ydmyg udtrykte gener 7,22 nøjagtigheden af microarray. Endelig microarray prober er forskellige i deres hybridiseringsegenskaber effektivitetsgevinster, der gør resultaterne mindre pålidelige, når man sammenligner de relative ekspressionsniveauer af forskellige transkripter inden for en prøve 23. Selv om RNA-Seq har mange fordele i forhold microarray, dens analyse af data er kompleks. Dette er en af grundene til, at mange forskere stadig bruger microarray stedet for RNA-Seq. Forskellige bioinformatiske værktøjer er nødvendige for RNA-Seq bearbejdning og analyse 24 data.
Blandt flere næste generation sekventering (NGS) platforme, 454, Illumina, solid og Ion Torrent er de mest anvendte dem. 454 var den første kommercielle NGS platform. I modsætning til de andre sekventering platformesåsom Illumina og SOLID, genererer 454 platform længere læse længde (gennemsnitligt 700 basen læser) 25. Længere læser er bedre for første karakterisering af transcriptiome på grund af deres højere samle effektivitet 25. Den største ulempe ved 454 platform er dens høje pris pr megabase af sekvens. Illumina og solid platforme generere læser med øgede tal og korte længder. Omkostningerne pr megabase af sekvensen er meget lavere end 454 platform. På grund af det store antal af kort læser for Illumina og faste platforme, dataanalyse er meget mere regnekraft. Prisen for instrumentet og reagenser til sekventering for Ion Torrent-platformen er billigere og sekventering er kortere 25. Dog er fejlprocenten og omkostningerne pr megabase af sekvens højere sammenlignet med Illumina og solide platforme. Forskellige platforme har deres egne fordele og ulemper og kræver forskellige metoder til dataanalyse. PLAtform bør vælges baseret på sekventering formål og tilgængeligheden af finansiering.
I dette papir, vi tager Illumina RNA-Seq platform som et eksempel. Vi brugte EML celle som et modelsystem til at undersøge de centrale lovgiverne i EML celle selvfornyelse og differentiering, og fremlagt en detaljeret metoder til RNA-Seq bibliotek konstruktion og dataanalyse for beregningsudtrykket niveau og roman udskrift detektion. Vi har vist i vores tidligere publikation, at RNA-seq studie i EML model system 2 når kombineret med funktionelle test (f.eks shRNA knockdown) give en kraftig tilgang i forståelsen af molekylære mekanisme af de tidlige stadier af hæmatopoietisk differentiering, og kan tjene som en model for analyse af celle selvfornyelse og differentiering i almindelighed.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. EML Cellekultur og Adskillelse af Lin-CD34 + Lin-CD34 celler under anvendelse Magnetic Cell Sorting System og fluorescens-aktiveret cellesortering Metode
- Fremstilling af babyhamsternyre (BHK) cellekulturmedium for stamcelle faktor samling:
- Kultur BHK-celler i DMEM medium indeholdende 10% FBS i 25 cm2 kolbe (tabel 1) ved 37 ° C, 5% CO 2 i en cellekultur inkubator.
- Når cellerne vokse til 80-90% sammenflydning, vaskes cellerne en gang med 10 ml PBS. Der tilsættes 5 ml 0,25% trypsin-EDTA-opløsning til monolaget og inkubere cellerne i 1-5 minutter ved stuetemperatur (RT), indtil cellerne er fritliggende.
- Pipette løsningen op og ned forsigtigt for at bryde op klumper af celler. Der tilsættes 5 ml komplet DMEM til kolben for at stoppe trypsin aktivitet. Indsamle cellerne ved centrifugering ved 200 xg i 5 minutter ved stuetemperatur.
- Fjern mediet og resuspender cellepelleten i 10 ml frisk BHK celledyrkningsmedium. Overfør 2 ml af cellesuspensionen fra trin 1.1.4 til en ny 75 cm 2 kolbe og tilsættes 48 ml frisk BHK celledyrkningsmedium til kolben.
- Dyrkning af BHK-celler i to dage og indsamle dyrkningsmediet. Passage mediet gennem et 0,45 um filter. Opbevar mediet i -20 ° C indtil yderligere anvendelse.
- EML cellekultur:
- Kultur EML celler (i suspension) i EML basisk medium indeholdende BHK-celle dyrkningsmedium (tabel 1) ved 37 ° C, 5% CO 2 i en cellekultur inkubator.
- Opretholde EML celler ved lav celletæthed (0,5-5 x 10 5 celler / ml) med den maksimale massefylde på mindre end 6 x 10 5 celler / ml. Opdele cellerne hver 2-3 dage i forholdet 1: 5. Passage EML celler forsigtigt og kassér kultur efter passage i 10 generationer.
- Nedbrydning af linjeforpligtede positive celler:
- Høste EML cellerne ved centrifugering ved 200 xg for 5 min og vaskes cellerne én gang med PBS. Indsamle cellerne ved centrifugering ved 200 xg i 5 minutter.
- Opblande cellerne med PBS og tælle celler med et hæmocytometer. Bestemme antistofkoncentration i den efterfølgende celle separation trin i forhold til antallet af celler (se instruktionerne, der tilbydes af udbyderen af cellen isolation system).
- Isoler afstamning negative (Lin-) celler ved hjælp af afstamning antistofcocktail (cocktail af biotinkonjugeret monoklonale antistoffer CD5, CD45R (B220), CD11, Anti-Gr-1 (Ly-6G / C), 7-4 og Ter-119 ) og en magnetisk cellesortering ifølge producentens anvisninger.
- Adskillelse af Lin-CD34 + og Lin-CD34-celler:
- Spin ned Lin- cellerne fra trin 1.3.3 ved 200 xg i 5 minutter. Resuspender cellepelleten med PBS og tælle cellerne med et hæmocytometer.
- Vask cellerne to gange med FACS buffer og pelletere cellerne ved 200 xgfor 5 min.
- Mærk fem 1,5 ml mikrocentrifugerør med nummer 1, 2, 3, 4, 5 hhv. Resuspender cellerne med 100 pi FACS buffer per 10 6 celler (10 6 celler pr rør).
- Tilsæt 1 ug Anti-Mouse CD34 FITC-antistof til rør 1 og rør 2 og bland rørene forsigtigt.
- Inkuber alle rør ved 4 ° C i 1 time i mørke.
- Tilføj 0,25 ug PE-konjugeret anti-Sca1 antistof og 20 pi APC-konjugeret Lineage Cocktail antistoffer mod røret 1, 0,25 ug PE-konjugeret anti-Sca1 antistof til røret 3, og 20 pi af APC-konjugerede Lineage Cocktail antistoffer til rør 4.
- Bland alle rørene forsigtigt og inkuberes cellerne ved 4 ° C i yderligere 30 minutter i mørke.
- Tilføj 300 pi FACS buffer til cellerne og spin ned cellerne ved 200 x g i 5 min.
- Vask cellerne med 500 pi FACS buffer til tre gange.
- Resuspender cellepelleten i 500 pi FACS buffer.
- Brug cellerne i rørene 2, 3, 4 og 5 for at oprette kompensation. Isoler Lin-SCA + CD34 + og Lin-SCA-CD34-celler i rør 1 ved hjælp af FACS Aria.
2. RNA Forberedelse og Library Construction for High-throughput sekventering
- Isolation, kvalitet analyse og kvantificering af RNA:
- Uddrag total RNA fra Lin-CD34 + og Lin-CD34-celler henholdsvis anvender TRIzol efter fremstiller »protokollen.
- Fjerne den forurenede DNA under anvendelse af deoxyribonuklease I (DNase I) efter fremstillingen protokol. Eventuelt opbevares RNA ved -80 ° C på dette trin til yderligere anvendelse.
- Vurdere kvaliteten af total RNA ved hjælp Bioanalyzer i henhold til instruktionerne, som leverandøren tilbyder. Brug RNA-prøve med RNA Integrity Number (RIN) pilsner end 9.
- Bibliotek Byggeri og high-throughput sekventering:
BEMÆRK: Denne protokol beskriver RNA-Seq hjælp Illumina platform. Forandre sekventering platforme, kræves der forskellige bibliotek fremstillingsmetoder.- Brug 0,1-4 ug af høj kvalitet total RNA per prøve for bibliotek forberedelse. Normalt 2 ug af total RNA kan ekstraheres fra 10 5 EML celler.
- Brug en RNA-sekventering prøveforberedelse for RNA oprensning og fragmentering, første og anden streng cDNA syntese, ende reparation, 3'-ender adenylering, adaptorligering og PCR-amplifikation, efter de detaljerede standardprocedurer fra udbyderens anvisninger.
- Vælg Positivt PolyA mRNA ved hjælp af oligo-dT magnetiske perler og opsplitte det mRNA.
- Udføre revers transskription under anvendelse af tilfældige primere til opnåelse af cDNA og derefter syntetisere den anden streng af cDNA til at generere dobbeltstrengede cDNA.
- Fjern de 3 'overhæng og udfylde 5'-fremspringene med DNA-polymerase. Adenylat 3'-ender for at forhindre cDNA-fragmenter fra ligere til hinanden.
- Tilføj multiplex indeksering adaptere til begge ender af dscDNA. Udfør PCR for berigelse af DNA-fragmenter.
- Mål A260 / A280 til at indhente oplysninger om koncentrationen af biblioteket ved hjælp af et spektrofotometer.
- Vurdere biblioteket kvalitet og måle størrelsesområdet af DNA-fragmenter ved hjælp af en Bioanalyzer.
3. Data Analysis
For reference af software, der bruges i denne del, se (Tabel 2).
- Datafilnavn behandling til downstream analyse:
- Konverter .bcl (base call-fil) fil til .fastq fil ved hjælp casava software (Illumina version 1.8.2).
- Fyr op på 'Terminal' i Linux-systemet. Gå til data mappe, der indeholder de data fil fra et Illumina HiSeq2000 sekventering maskine. Antag resultatet mappen er "NASboy1 / JiaqianLabData / HiSeq_RUN / 2013_07_11 / 130627_SN860_0309_A_2013-166_H0PW9ADXX / ', typei kommandoen i figur S1A og indtaste data mappen.
- Installer casava 1.8.2 i Linux-systemet. Antag outputfolder er »unaligned«, bruge kommandoen i figur S1B at forberede konfigurationsfil til konvertering. Brug indstillingen --fastq-cluster-count 0 til kun at sikre en .fastq filen er oprettet for hver prøve. Den genererede .fastq fil er i .gz format. Unzip det for downstream analyse (figur S1B).
- Efter »unaligned 'folder er blevet genereret, gå til' unaligned 'mappe (figur S1c).
- Brug kommandoen i fig S1D at begynde konvertering af processen. Den "-j 'parameter leverer cpu nummer, der vil blive anvendt.
- Når systemet er færdig konvertere processen, gå til resultatet mappe under 'unaligned' mappe (figur S1E).
- Brug kommandoen i figur S1F </ Strong> at dekomprimere .fastq.gz fil i .fastq fil under hver prøve mappe.
- Konverter .bcl (base call-fil) fil til .fastq fil ved hjælp casava software (Illumina version 1.8.2).
- Detect nye udskrifter og evaluere udtrykket niveau ved hjælp Tuxedo Suite 26:
- Kortlæg parret ende RNA-Seq læser til referencen musegenomet (UCSC versionen MM9, opnået fra http://cufflinks.cbcb.umd.edu/igenomes.html ) ved hjælp Tophat software (version 1.3.3) 27, som bruger Den Bowtie læse mapper (version 0.12.7) 28. Tophat leveres med "-ingen-nye-juncs" mulighed for at forbedre estimering nøjagtighed udtryk niveau.
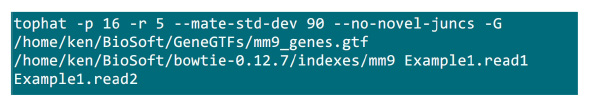
- Sæt .fastq filer i en mappe, hvor kortlægningen vil blive gennemført. Antag at der er 2 .fastq filer (omdøbe til Example1.read1, Example1.read2) for en parret ende sekventering prøve, skal du bruge kommandoen i figur S2 at gøre kortlægningen (justere parametrene i overensstemmelse med systemet indstilling).Den "-p" parameteren leverer cpu nummer, der vil blive brugt. De "-r" og "-mate-STD-dev" parametre kan fås fra biblioteket QC eller udledes af en delmængde af linie læser (Figur S2).
- Saml kortlagt læser i RNA-transkripter ved hjælp af Manchetknapper software (version 1.3.0) 29. Kør Manchetknapper vha annotationsfil af kendte gener (samme .gtf fil, der bruges af Tophat) og .bam fil produceret med Tophat.
- Efter Tophat færdig med at køre i den samme mappe, skal du bruge kommandoen i figur S3A at køre manchetknapper at konstruere transkriptom og skøn udskrift udtryk niveau. Den "mm9_repeatMasker.gtf« og genomsekvens filer i mappen 'GenomeSeqMM9' kan fås fra UCSC Genome Browser.
- De resulterende genes.expr og transcripts.expr filer indeholder udtrykket værdi af gener og udskrifter (isoformer). Kopier og indsætfilindholdet til en Excel-fil og manipulere med regneark (figur S3B).
- Brug kommandoen i figur S3C at sammenligne den resulterende »transcripts.gtf 'filen til referencen' mm9_genes.gtf 'fil for at identificere nye udskrifter.
- Den resulterende .tmap fil indeholder sammenligningen resultat. Kopier og indsæt filindholdet til en Excel-fil og manipulere med regnearksprogram. Udskrifter med klasse kode 'u' kan betragtes som 'nye' i forhold til referencen .gtf fil, (Figur S3D).
BEMÆRK: For downstream analyse bekvemmelighed, indstille FPKM værdier til 0,1, hvis værdierne er under 0,1.
BEMÆRK: Trin 3.2.3 - 3.2.6 er valgfrit for dem, der ønsker at forbedre nøjagtigheden af nye udskrifter 'udtryk skøn. Dette vil tage meget længere tid, fordi kortlægning og transkriptom konstruktion skal være run mere end én gang.
- Kør Tophat at bruge standard parametre og derefter køre manchetknapper til genereret .gtf fil ved hjælp af kommandoen i figur S3E.
- Sammenlign resulterende .gtf filen til referencen genom .gtf fil ved hjælp af kommandoen i figur S3F.
- Parse resulteret .tmap fil som beskrevet i trin 3.2.2.4. Kopier og indsæt filindholdet til en Excel-fil og manipulere med regnearksprogram. Udskrifter med klasse kode 'u' kan betragtes som 'nye' i forhold til referencen .gtf fil forudsat.
- Efter trin 3.2.5, er der en .combined.gtf fil i mappen, som kan bruges som reference .gtf fil. En anden kørsel af Tophat og manchetknapper kan udføres som beskrevet i trin 3.2.1 og 3.2.2 at opnå en mere nøjagtig FPKM estimering af nye transkripter.
- Kortlæg parret ende RNA-Seq læser til referencen musegenomet (UCSC versionen MM9, opnået fra http://cufflinks.cbcb.umd.edu/igenomes.html ) ved hjælp Tophat software (version 1.3.3) 27, som bruger Den Bowtie læse mapper (version 0.12.7) 28. Tophat leveres med "-ingen-nye-juncs" mulighed for at forbedre estimering nøjagtighed udtryk niveau.
- Detect differentially udtrykte gener ved hjælp DESeq pakke 30.
- Inputtet af DESeq er en rå læste tæller bordet. For at opnå en sådan tabel, skal du bruge htseq-count script distribueres med HTSeq Python pakke, som kan downloades fra HTSeq hjemmeside ( http://www-huber.embl.de/users/anders/HTSeq/doc/count.html ) .
- Sørg for, at samtools, Python og htseq-count programsare installeret i systemet. Opnå rå read tæller numre fra Tophat output ved hjælp af kommandoen i figur S4A.
- Forbered 'Raw_Count_Table.txt «,» ExperimentDesign.txt' filer ved hjælp af Excel. Kopier og gem det indhold i .txt-format for DESeq R-pakken (Figur s4b).
- Installer R program i systemet. I terminalen, viste type »R« og tryk ENTER.A skærm meddelelse appearas i figur S4C.
- Læs 'Raw_Count_Table.txt ',' ExperimentDesign.txt 'i R ved hjælp af kommandoen i figur S4D.
- Load DESeq pakken ved hjælp af kommandoen i figur S4E.
- Faktorisere forhold i R (figur S4F).
- Brug kommandoen i fig S4G at køre negative binominal test på den normaliserede tæller tabel.
- Brug kommandoen i figur S4H til output væsentlig forskel udtrykte gener i en .csv-fil.
- Inputtet af DESeq er en rå læste tæller bordet. For at opnå en sådan tabel, skal du bruge htseq-count script distribueres med HTSeq Python pakke, som kan downloades fra HTSeq hjemmeside ( http://www-huber.embl.de/users/anders/HTSeq/doc/count.html ) .
- Opslag transkriptionsfaktorer «(TFS) FPKM værdier på tværs af prøver ved hjælp af Excel. Intersect DE gen bord og TF'er tabel. Gener tilhører både skemaet differentielt udtrykte transkriptionsfaktorer.
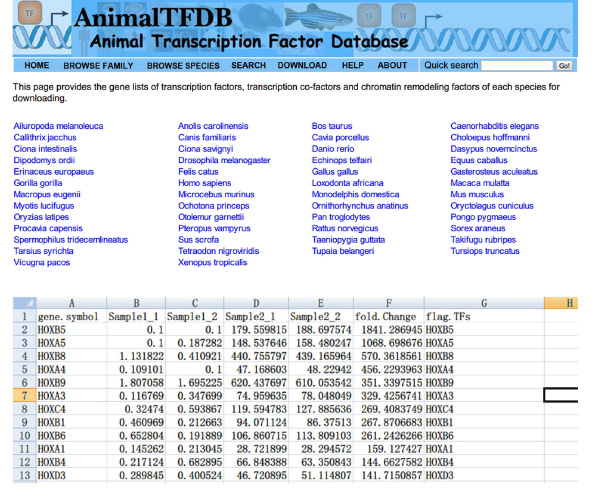
- Gå til webstedet http://www.bioguo.org/AnimalTFDB/download.php og hente transkriptionsfaktorer. Derefter opslag DE transkriptionsfaktorer i Excel (< strong> Figur S5).
- Generering .bigwig fil til UCSC genom browser visualisering.
- Hent 'bedtools' softwarepakke fra hjemmesiden https://github.com/arq5x/bedtools2 og installere software i systemet 31. Hent UCSC værktøjernes bedGraphToBigWig 'fra hjemmesiden http://hgdownload.cse.ucsc.edu/admin/exe/ og installere software i systemet.
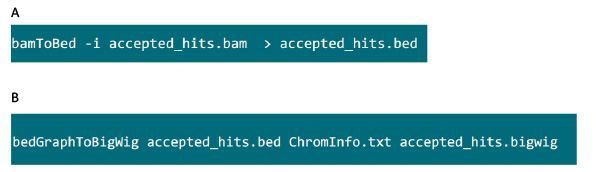
- I mappen med .bam fil, bruge kommandoen i figur S6A at konvertere .bam fil genereret af Tophat i .bed fil.
- Når .bed fil er produceret, skal du bruge kommandoen i figur S6B at generere .bigwig fil. Filen 'ChromInfo.txt "kan fås fra følgende url:Arget = "_blank"> http://hgdownload.cse.ucsc.edu/goldenPath/mm9/database/chromInfo.txt.gz.
- Observere en brugerdefineret spor på UCSC Genome Browser. Der henvises til hjemmesiden http://genome.ucsc.edu/goldenPath/help/customTrack.html om hvordan du får vist en brugerdefineret spor ved hjælp af UCSC genom browser.

Figur S1: Konvertering .bcl fil til .fastq fil ved hjælp casava software.

Figur S2: Mapping læser at referere genom ved hjælp Tophat.
Figur S3: Påvisning af nye udskrifter og udtryk estimering.

Figur S4: Calling forskellen udtrykt gen ved hjælp DESeq pakke.

Figur S5: Identifikation af differentielt udtrykte transkriptionsfaktorer.

Figur S6: Konvertering kortlægning resultat for data visualisering.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
For at analysere differentielt udtrykte gener i Lin-CD34 + og Lin-CD34- EML celler, vi brugte RNA-Seq teknologi. Figur 1 viser arbejdsgangen af procedurerne. Efter isolering af linjeforpligtede negative celler ved magnetisk cellesortering, vi adskilt Lin-SCA + CD34 + og Lin-SCA-CD34-celler under anvendelse af FACS Aria. Lin-beriget EML-celler blev farvet med anti-CD34, anti-Sca1 og afstamning cocktail antistoffer. Kun Lin celler blev gated for analyse af Sca1 og CD34-ekspression. To populationer (SCA + CD34 + og SCA-CD34- EML celler) kunne observeres ved FACS-analyse (figur 2) 6.
Efter celleseparering, vi ekstraheres totalt RNA fra CD34 + og CD34-celler henholdsvis og analyseret kvaliteten af RNA. Nøjagtigheden af RNA-Seq data i høj grad afhænger af kvaliteten af RNA-Seq bibliotek og kvaliteten af total RNA er afgørende for fremstilling af en høj kvalitet bibliotek. Høj kvalitet RNA-prøve skal have en OD 260/280 værdi mellem 1.8 og 2,0. Ud over ved spektrofotometri blev RNA kvalitet yderligere vurderes med større nøjagtighed ved Bioanalyzer. Figur 3 viser et resultat af en høj kvalitet RNA-prøve med RIN lig med 9,4. Høj kvalitet total RNA-prøve med RIN værdi større end 9 blev anvendt til mRNA-ekstraktion og efterfølgende bibliotekskonstruktionsteknikker procedurer.
Ribosomale RNA er den mest udbredte form for RNA i cellen. I øjeblikket to hovedstrategier, udtømning af rRNA eller positivt valg af polyadenyleret mRNA (poly-A mRNA), anvendes til berigelse af mål-RNA før bibliotek konstruktion. Ikke polyadenylerede RNA-arter går tabt under udvælgelsen af poly-A mRNA. I modsætning hertil kunne rRNA depletion metoder såsom RiboMinus bevare ikke polyadenylerede RNA-arter. Formålet med vores undersøgelse er at kigge efter differentielt udtrykte kodning gener i to celletyper, således vi brugt poly-A mRNA udvælgelsesmetode til berigelse af mål-RNA'er før biblioteket entrektion. Når bibliotek konstruktion var færdig, blev størrelsen af DNA-fragmenter i biblioteket kontrolleres før sekvensering ved brug Bioanalyzer. Figur 4 viser en god kvalitet bibliotek med fragment størrelse toppe ved ca. 300 bp.
I det efterfølgende trin, blev biblioteket underkastes high-throughput sekventering. I princippet vil længere læse længden være nyttigt for read mapping. Det kan reducere sandsynligheden for, at læsning er kortlagt til flere steder på grund af ligheden mellem duplikerede gener eller gen-familiemedlemmer. Som par-endesekventering sekvenser er fra begge ender af fragmenterne skal læse længde vælges mindre end halvdelen af den gennemsnitlige fragmenter længde. Hvis hovedformålet med eksperimentet er at måle ekspressionsniveauet i stedet for at konstruere afskrift struktur, single-ende læse (75 eller 100 bp) kan reducere omkostningerne uden at miste for meget information. Forbundne-endesekventering er mere nyttigt for udskrift struktur konstruktion og korterelæse længde kan anvendes til at reducere omkostningerne. Bestemt, når der er tilstrækkelig midler til rådighed, der er længere læse længde foretrækkes.
For differentiel ekspression analyse, der er mange andre end DESeq alternative algoritmer. Der er også en inkluderet i manchetknapper pakke opkaldt cuffdiff 32. DESeq er en af de mest udbredte tæller baseret DE genanalyse algoritmer. DESeq metode er baseret på en velkarakteriseret statistik model - negative binomialfordeling. Det er vores erfaring, DESeq er mere stabil sammenlignet med cuffdiff. Tidlige versioner af cuffdiff giver ofte markant forskellige numre af De gener. Derfor brugte vi DESeq for DE analyse her.
Fordi transkriptionsfaktorer er afgørende for celle skæbne beslutsomhed, fokuserede vi på den markant differentielt udtrykte transskriptionsfaktorerne 33. TFS ændrede> 1,5 gange mellem Lin-CD34 + og Lin-CD34- blev fundet og er vist på Heatmap (Figure 5) 2. Især det relative ekspressionsniveau af Tcf7 i Lin-CD34 + -celler er mere end 100 gange højere end i Lin-CD34-celler. Således Tcf7 blev valgt til yderligere chip sekventering (Kromatin- Immunopræcipitation og sekventering) analyse og funktionel test for at bekræfte Tcf7 's funktion i reguleringen af EML celle selvfornyelse og differentiering 2.

Figur 1:. Arbejdsgang af procedurerne Lin-CD34 + Lin-CD34-celler blev separeret ved magnetisk celleseparering, og fluorescens-aktiveret cellesortering metode. Total RNA blev ekstraheret efterfulgt af mRNA oprensning og bibliotek konstruktion. Efter analyse af biblioteket kvalitet, blev prøverne udsat for high throughput sekventering. Data blev analyseret og differentielt udtrykt transkriptionsfaktorer blev identificeret.

Figur 2: Adskillelse af Lin-CD34 + og Lin-CD34- EML celler 6 Lin- EML celler blev beriget ved magnetisk cellesortering.. Lin- celler blev farvet med anti-CD34, anti-Sca1 og afstamning blandingen antistoffer. Lin celler blev gated for ekspression af CD34 og Sca1. Lin-CD34 + SCA + og Lin-CD34-SCA- EML cellepopulationer blev sorteret.

Figur 3:. En repræsentant for høj kvalitet total RNA-prøve var kvaliteten af total RNA vurderet af Bioanalyzer. RNA Integrity Number er 9,4 (FU fluorescens Units).

Figur 4:. Fragmenter størrelse vifte af Forbundne-End biblioteket DNA størrelsesfordelingen af biblioteket blev analyseret ved anvendelse Bioanalyzer. De fleste fragmenter er inden for størrelsesområdet 250-500 bp.

Figur 5:. Differentielt udtrykte transkriptionsfaktorer (> 1,5 fold) mellem Lin-CD34 + celler og Lin-CD34-celler 2 For hver celletype blev to uafhængige forsøg udført. Opregulerede gener er angivet som rød farve og ned-regulerede gener er angivet som grøn farve.
Tabel 1: Buffere og Cell dyrkningsmedier.
| Software | Usage | Henvisning | |||
| Bowtie 1.2.7 | Anvendes af Tophat til kortlægning | [28] | |||
| Tophat 1.3.3 | Mapping læser tilbage til henvisning genom | [27] | |||
| Manchetknapper 1.3.0 | Transkripter konstruktionen og ekspressionen estimering | [29] | |||
| DESeq 1.16.0 | Differentiel ekspression analyse | [30] | Bedtools 2,18 | Konverter .bam fil i .bed fil | [31] |
| bedGraphToBigWig | Konverter .bed fil til .bigwig fil | http://genome.ucsc.edu/ |
Tabel 2: Liste over software til dataanalyse.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Mammalian transkriptom er meget kompleks 34-38. RNA-Seq teknologi spiller en stadig vigtigere rolle i undersøgelser af transkriptom analyse roman udskrifter afsløring og enkelt nukleotid variation opdagelse etc. Det har mange fordele frem for andre metoder til genekspression analyse. Som nævnt i indledningen, det overvinder de hybridiseringsbetingelser artefakter microarray og kan anvendes til at identificere hidtil ukendte transkripter de novo. En begrænsning af RNA-sekventering er relativ kort læse længde sammenligne Sanger-sekventering. Men med hurtig forbedring af sekventering teknologi, læse længde er konstant stigende. I dette papir, vi giver detaljerede metoder til at bruge denne teknologi til at identificere potentielle vigtige regulatorer i muse EML celle selvfornyelse og differentiering.
Det første afgørende skridt for denne protokol er EML cellekultur. Selv EML er en hæmatopoietisk precursor cellelinje, og det kan væreformeret i store mængder med SCF. Dyrkningen tilstand EML celler kræver mere opmærksomhed, end de sædvanlige immortaliserede cellelinier. Cellerne skal fodres og passeret på en regelmæssig basis med blid operation; ellers cellerne kunne ændre i deres egenskaber af selvfornyelse og differentiering og undergå celledød. Som det første trin efter indsamling nok celler isolerede vi afstamning negative celler under anvendelse af en magnetisk cellesortering system. Så vi adskilt CD34 + og CD34-celler under anvendelse af fluorescens-aktiveret celle-sortering. De EML celler normalt passeres mindre end 10 generationer, før du bruger for RNA ekstraktion og antallet af CD34 + og CD34-celler skal være ens efter separation. Hvis de to populationer varierer meget i celle nummer, er det tilrådeligt at kassere kultur og re-tø andet rør celle materiel til kulturen.
Efter separation af CD34 + og CD34- celler blev totalt RNA-ekstraktion udføres endnu et vigtigt skridt for denne mudy. Højkvalitets RNA er grundlaget for konstruktionen af høj kvalitet bibliotek, som lover nøjagtigheden af sekventering data. I denne kritiske fase, bør enhver kontakt med RNase undgås. Alle reagenser skal RNase-frit. Det er vigtigt at bære handsker på alle tidspunkter mens håndtering RNA. Højkvalitets RNA-prøven har en OD 260/280 værdi mellem 1,8 og 2,0. Ved afhentning af vandig fase indeholdende RNA, være omhyggelig med ikke at bære nogen organiske fase med RNA-prøven. Eventuelle resterende organiske opløsningsmidler, såsom phenol eller chloroform i RNA vil resultere i en OD260 / 280-værdi lavere end 1,65. Hvis OD260 / 280-værdi er lavere end 1,65, udfælde RNA igen med ethanol. Efter vask med 75% ethanol, ikke overdry RNA pellet. Tørring RNA pellet fuldstændigt vil påvirke opløseligheden af RNA og føre til lavt udbytte af RNA.
Det næste afgørende skridt for denne protokol er bibliotek forberedelse. Efter total RNA-ekstraktion, et trin til anvendelse af DNase til fjernelse af forurenet DNA is anbefales, idet DNA-kontaminering kan resultere i forkert vurdering af mængden af total RNA anvendes. Det anbefales at udføre nedstrøms procedure umiddelbart efter RNA-isolering, idet efter lang tids opbevaring og frysetørring procedure, vil RNA nedbrydes til en vis grad. Hvis de efterfølgende skridt efter RNA isolation ikke kan udføres straks, skal den opbevares RNA i -80 ° C. Før der anvendes total RNA til mRNA oprensning og cDNA-syntese, bør kvaliteten altid kontrolleres. Høj kvalitet RNA kan anvendes til biblioteket forberedelse. Brug af lav kvalitet eller forringet RNA kan føre til en overrepræsentation af 3'-ender. Før sekventering blev biblioteket kvalitet vurderes for at sikre maksimal sekventering effektivitet.
I dataanalyse del, efter at have udført en kørsel af manchetknapper uden en henvisning transkriptom, vi kombinerede de nye udskrifter med kendte udskrifter til at danne en henvisning .gtf fil og køre Tophat og manchetknapper for anden gang.Denne to-run procedure anbefales, da dette giver mere præcis FPKM estimering end at køre én gang. Efter dataanalyse blev differentielt udtrykte gener identificeret. Downstream eksperimenter kan udføres for at validere funktionen af gener in vitro og in vivo. I vores tidligere publikation 2, valgte vi de væsentligt differentielt udtrykte transkriptionsfaktorer og identificeret genomet bindingssted af disse faktorer ved at udføre kromatin immunofældning og sekventering (chip-Seq). Desuden anvendes vi shRNA knockdown assay til at teste den funktionelle virkning af Tcf7. Vi fandt, at i Tcf7 knockdown celler, up-regulerede gener var generne højt beriget i CD34-celler, mens nedregulerede gener fandtes at være væsentligt beriget med CD34 + -celler. Derfor genekspressionsprofilen af Tcf7 knockdown-celler forskydes mod et delvist differentieret CD34- state.Overall anvendelse EML celle som et modelsystemkombineret med RNA-sekventering teknologi og funktionelle assays, vi identificeret og bekræftet Tcf7 som en vigtig regulator af EML celle selvfornyelse og differentiering.
Subscription Required. Please recommend JoVE to your librarian.
Materials
| Name | Company | Catalog Number | Comments |
| Antibiotic-Antimycotic | Invitrogen | 15240-062 | BHK cell culture |
| Anti-Mouse CD34 FITC | eBioscience | 11-0341-81 | FACS sorting |
| Anti-Mouse Ly-6A/E (Sca-1) PE | eBioscience | 12-5981-81 | FACS sorting |
| APC Mouse Lineage Antibody Cocktail | BD Biosciences | 558074 | FACS sorting |
| BD FACSAria Cell Sorter | BD Biosciences | Special offer sysmtem | FACS sorting |
| Corning™ Cell Culture Treated Flasks 75 cm2 | Corning incorporated | 430641 | Cell culture |
| Corning™ Cell Culture Treated Flasks 25 cm2 | Corning incorporated | 430639 | Cell culture |
| Deoxyribonuclease I, Amplification Grade | Invitrogen | 18068-015 | Library preparation |
| DMEM | Invitrogen | 11965-092 | BHK cell culture |
| DPBS | Gibco | 14190 | Cell culture |
| HI FBS | Invitrogen | 16140071 | BHK cell culture |
| Horse Serum | Invitrogen | 16050-122 | EML cell culture |
| IMDM | HyClone | SH30228.02 | EML cell culture |
| L-Glutamine | Invitrogen | 25030-081 | Cell culture |
| Lineage Cell Depletion Kit, mouse | Miltenyi Biotec | 130-090-858 | Isolation of lineage negative cells |
| NanoVue Plus spectrophotometer | GE Healthcare | 28-9569-62 | Quality control |
| Thermo Scientific™ Napco™ 8000 Water-Jacketed CO2 Incubators | Thermo Scientific | 15-497-002 | Cell culture |
| Penicillin-Streptomycin | Invitrogen | 15140-122 | EML cell culture |
| TRIzol® Reagent | Invitrogen | 15596-018 | RNA exraction |
| TruSeq™ RNA Sample Prep Kit v2 -Set B (48 rxn) | Illumina | RS-122-2002 | Library preparation |
| 2100 Electrophoresis Bioanalyzer Instrument | Agilent | G2939AA | Quality control |
| 0.25% Trypsin-EDTA | Gibco | 25200 | Cell culture |
| 0.45 µm Syringe Filters | Nalgene | 190-2545 | Cell culture |
References
- Chambers, S. M., Goodell, M. A. Hematopoietic stem cell aging: wrinkles in stem cell potential. Stem Cell Rev. 3, 201-211 (2007).
- Wu, J. Q., et al. Tcf7 is an important regulator of the switch of self-renewal and differentiation in a multipotential hematopoietic cell line. PLoS genetics. 8, (2012).
- Ye, Z. J., et al. Complex interactions in EML cell stimulation by stem cell factor and IL-3. Proceedings of the National Academy of Sciences of the United States of America. 108, 4882-4887 (2011).
- Tsai, S., Bartelmez, S., Sitnicka, E., Collins, S. Lymphohematopoietic progenitors immortalized by a retroviral vector harboring a dominant-negative retinoic acid receptor can recapitulate lymphoid, myeloid, and erythroid development. Genes Dev. 8, 2831-2841 (1994).
- Weiler, S. R., et al. D3: a gene induced during myeloid cell differentiation of Linlo c-Kit+ Sca-1(+) progenitor cells. Blood. 93, 527-536 (1999).
- Ye, Z. J., Kluger, Y., Lian, Z., Weissman, S. M. Two types of precursor cells in a multipotential hematopoietic cell line. Proc Natl Acad Sci U S A. 102, 18461-18466 (2005).
- Wang, Z., Gerstein, M., Snyder, M. RNA-Seq: a revolutionary tool for transcriptomics. Nature reviews. Genetics. 10, 57-63 (2009).
- Chu, Y., Corey, D. R. RNA sequencing: platform selection, experimental design, and data interpretation. Nucleic acid therapeutics. 22, 271-274 (2012).
- Hornett, E. A., Wheat, C. W. Quantitative RNA-Seq analysis in non-model species: assessing transcriptome assemblies as a scaffold and the utility of evolutionary divergent genomic reference species. BMC genomics. 13, 361 (2012).
- Eswaran, J., et al. RNA sequencing of cancer reveals novel splicing alterations. Scientific reports. 3, 1689 (2013).
- Wang, E. T., et al. Alternative isoform regulation in human tissue transcriptomes. Nature. 456, 470-476 (2008).
- Wu, J. Q., et al. Dynamic transcriptomes during neural differentiation of human embryonic stem cells revealed by short, long, and paired-end sequencing. Proceedings of the National Academy of Sciences of the United States of America. 107, 5254-5259 (2010).
- Loraine, A. E., McCormick, S., Estrada, A., Patel, K., Qin, P. RNA-seq of Arabidopsis pollen uncovers novel transcription and alternative splicing. Plant physiology. 162, 1092-1109 (2013).
- Edgren, H., et al. Identification of fusion genes in breast cancer by paired-end RNA-sequencing. Genome biology. 12, 6 (2011).
- Ilott, N. E., Ponting, C. P. Predicting long non-coding RNAs using RNA sequencing. Methods. 63, 50-59 (2013).
- Sun, L., et al. Prediction of novel long non-coding RNAs based on RNA-Seq data of mouse Klf1 knockout study. BMC bioinformatics. 13, 331 (2012).
- Luo, S. MicroRNA expression analysis using the Illumina microRNA-Seq Platform. Methods in molecular biology. 822, 183-188 (2012).
- Bolduc, F., Hoareau, C., St-Pierre, P., Perreault, J. P. In-depth sequencing of the siRNAs associated with peach latent mosaic viroid infection. BMC molecular biology. 11, 16 (2010).
- Chepelev, I., Wei, G., Tang, Q., Zhao, K. Detection of single nucleotide variations in expressed exons of the human genome using RNA-Seq. Nucleic acids research. 37, 106 (2009).
- Djari, A., et al. Gene-based single nucleotide polymorphism discovery in bovine muscle using next-generation transcriptomic sequencing. BMC genomics. 14, 307 (2013).
- Murphy, D. Gene expression studies using microarrays: principles, problems, and prospects. Advances in physiology education. 26, 256-270 (2002).
- Chen, K., et al. RNA-seq characterization of spinal cord injury transcriptome in acute/subacute phases: a resource for understanding the pathology at the systems level. PLoS one. 8, 72567 (2013).
- Marioni, J. C., Mason, C. E., Mane, S. M., Stephens, M., Gilad, Y. RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome research. 18, 1509-1517 (2008).
- Ramskold, D., Kavak, E., Sandberg, R. How to analyze gene expression using RNA-sequencing data. Methods in molecular biology. 802, 259-274 (2012).
- Glenn, T. C. Field guide to next-generation DNA sequencers. Mol Ecol Resour. 11, 759-769 (2011).
- Trapnell, C., et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nature protocols. 7, 562-578 (2012).
- Trapnell, C., Pachter, L., Salzberg, S. L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 25, 1105-1111 (2009).
- Langmead, B., Trapnell, C., Pop, M., Salzberg, S. L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome biology. 10, 25 (2009).
- Trapnell, C., et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature. 28, 511-515 (2010).
- Anders, S., Huber, W. Differential expression analysis for sequence count data. Genome biology. 11, 106 (2010).
- Quinlan, A. R., Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 26, 841-842 (2010).
- Cheranova, D., et al. RNA-seq analysis of transcriptomes in thrombin-treated and control human pulmonary microvascular endothelial cells. J Vis Exp. , (2013).
- Zhang, H. M., et al. AnimalTFDB: a comprehensive animal transcription factor database. Nucleic acids research. 40, 144-149 (2012).
- Wu, J. Q., et al. Systematic analysis of transcribed loci in ENCODE regions using RACE sequencing reveals extensive transcription in the human genome. Genome Biol. 9, 3 (2008).
- Wu, J. Q., et al. Large-scale RT-PCR recovery of full-length cDNA clones. Biotechniques. 36, 690-696 (2004).
- Wu, J. Q., Shteynberg, D., Arumugam, M., Gibbs, R. A., Brent, M. R. Identification of rat genes by TWINSCAN gene prediction, RT-PCR, and direct sequencing. Genome Res. 14, 665-671 (2004).
- Dewey, C., et al. Accurate identification of novel human genes through simultaneous gene prediction in human, mouse, and rat. Genome Res. 14, 661-664 (2004).
- Wu, J. Characterize Mammalian Transcriptome Complexity. , LAP Lambert Academic Publishing. (2011).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}