

Summary
RNA-sıralama ve biyoinformatik analizler fare EMLcells Lin-CD34 + ve Lin-CD34 alt gruplarda anlamlı ve farklı şekilde ifade transkripsiyon faktörleri tanımlamak için kullanılmıştır. Bu transkripsiyon faktörleri kendini sürekli yenileyen Lin-CD34 + ve kısmen farklılaşmış Lin-CD34 hücreleri arasındaki geçiş belirlenmesinde önemli rol oynayabileceğine.
Abstract
Hematopoetik kök hücreler (HSC), lösemi ve lenfoma gibi birçok hastalıkta hastanın hematopoetik sistemi yeniden inşa etmek nakli tedavisi için klinik olarak kullanılmaktadır. HSC kendini yenileme ve farklılaşmasını kontrol eden mekanizmaların tanıtılması araştırma ve klinik kullanım için HKH'lerin uygulaması için önemlidir. Bununla birlikte, bunun nedeni, in vitro proliferasyonu edemedikleri için HKH'lerin büyük miktarda elde etmek mümkün değildir. Bu engeli aşmak için, bu çalışma için, bir model sistem olarak, bir fare kemik iliği türevi hücre hattı, EML (eritroid, miyeloid ve lenfositik) hücre hattı kullanılmıştır.
RNA sıralaması (RNA-Dizi) giderek artan bir gen ifade çalışmaları için mikrodizisi değiştirmek için kullanılmıştır. Biz burada EML hücre kendini yenileme ve farklılaşma düzenlenmesinde potansiyel anahtar faktörleri araştırmak için RNA-Sıra teknolojisini kullanarak ayrıntılı bir yöntem rapor. Bu yazıda verilen protokol üç bölüme ayrılmıştır. Ilk part nasıl kültür EML hücreleri ve ayrı Lin-CD34 + ve Lin-CD34 hücreleri açıklar. Protokolün ikinci bölümü, toplam RNA hazırlama ve yüksek sekanslama için daha sonra kütüphane yapımı için ayrıntılı prosedürler bulunmaktadır. Son bölüm RNA-Dizi veri analizi için yöntem açıklanır ve Lin-CD34 + ve Lin-CD34 hücreleri arasındaki farklı şekilde ifade transkripsiyon faktörleri belirlemek için verilerin nasıl kullanılacağını açıklar. En önemli ölçüde farklı olarak ifade transkripsiyon faktörleri EML hücre kendini yenileme ve farklılaşmasını kontrol eden potansiyel kilit düzenleyiciler olduğu tespit edildi. Bu yazının tartışma bölümünde, bu deney başarılı performans için önemli adımlar vurgulayın.
Özetle, bu kağıt EML hücrelerinde kendini yenileme ve farklılaşma potansiyeli regülatörleri belirlemek için RNA-Sıra teknolojisini kullanarak bir yöntem sunar. Belirlenen anahtar faktörler in vitro ve i aşağı fonksiyonel analize tabi tutulurlarin vivo koşullarda.
Introduction
Hematopoetik kök hücreler erişkin kemik iliği niş ağırlıklı olarak ikamet nadir kan hücreleridir. Bunlar, kan yeniden doldurmak için gereken hücreleri ve immün sistem 1 üretimi için sorumludur. Kök hücrelerin bir tür olarak, HSC kendini yenileme ve farklılaşma kabiliyetine sahip bulunmaktadır. HKH'lerin kaderi kararını kontrol mekanizmaların tanıtılması, kendini yenileme veya farklılaşma ya doğru, kan hastalığı araştırmalar ve klinik kullanım için 2 HKH'lerin manipülasyon değerli rehberlik sunacak. Araştırmacılar tarafından karşılaşılan bir sorun, HSC muhafaza ve çok sınırlı bir ölçüde, in vitro olarak genişletilebilir olabilir; soylarına büyük çoğunluğu kısmen kültür 2 ayrıştırılmıştır.
Genom ölçeğinde kendini yenileme ve farklılaşma süreçlerini kontrol tuşuna regülatörleri tanımlamak için, biz bir model sistem olarak bir fare ilkel hematopoetik öncül hücre hattını EML kullanılır. Thhücre hattı fare kemik iliği 3,4 türetilmiştir edilir. Farklı büyüme faktörleri ile verildiğinde, EML hücrelerin in vitro 5 eritrosit, miyeloid ve lenfoid hücrelerine dönüştürmek olabilir. Önemli olarak, bu hücre hattı kültürü ortamı bunların multipotentiality tutma hala kök hücre faktörü (SCF) ihtiva eden ve büyük miktarda çoğaltılabilir. EML hücreler kendini sürekli yenileyen Lin-SCA + CD34 + alt popülasyonlar ayrılır ve kısmen yüzey belirteçleri CD34 ve SCA 6 dayalı Lin-SCA-CD34 hücreleri ayırt edilebilir. Kısa vadeli HKH'lerin, SCA + CD34 + hücrelerde de benzer kendini yenileme edebiliyoruz. Hızla Lin-SCA + CD34 + ve Lin-SCA-CD34 hücre karışık bir nüfus yeniden ve 6 çoğalmaya devam edebilirsiniz SCF, Lin-SCA + CD34 + hücre ile tedavi edildiğinde. Popülasyonlar morfolojik olarak benzer ve c-kit mRNA ve protein 6 benzer seviyelerine sahiptir. Lin-SCA-CD34 hücreleri, IL-3 yerine SCF3 ihtiva eden ortam içinde yayılan yeteneğine sahiptirler. Unveiling hematopoiez sırasında erken gelişimsel geçiş hücresel ve moleküler mekanizmaların daha iyi anlaşılmasını sunacak EML hücre kaderinin kararda kilit düzenleyiciler.
Kendini sürekli yenileyen Lin-SCA + CD34 + ve kısmen farklılaşmış Lin-SCA-CD34 hücreleri arasındaki yatan moleküler farklılıkları araştırmak amacıyla, farklı şekilde ifade genleri tanımlamak için RNA-seg kullanılır. Transkripsiyon faktörleri hücre kaderini belirlemede önemli olduğu gibi, özellikle biz, transkripsiyon faktörleri odaklanmak. RNA-Seq profil ve genom 7,8 transkripsiyonu RNA'lar ölçmek için (NGS) teknolojileri yeni nesil dizileme yeteneklerini kullanan yeni geliştirilmiş bir yaklaşımdır. Kısaca, total RNA, poli-A, ilk template.The RNA şablonu ile sonra ters transkriptazı kullanılarak cDNA'ya dönüştürülür olarak seçilir ve parçalanmış. CDNA kütüphanesinin oluşturulması için sağlam olmayan bozulmuş RNA kullanılarak, tam uzunlukta RNA transkriptlerinin haritasını yapmak için önemlidir. Amaçlarlasekanslama poz, belirli bir adaptör dizileri cDNA'nm her iki ucuna eklenir. Daha sonra, bir çok durumda, cDNA molekülleri, PCR ile amplifiye edilmiş ve yüksek verimli bir şekilde dizildi.
Sekanslama sonra, elde edilen bir bir referans genom ve transkriptom veritabanına hizalanabilir okunur. Sayısı referans gen haritası sayılır ve bu bilgi gen salgılama seviyesini tahmin için kullanılabileceğini okur. Ayrıca sigara model organizmaların 9 transcriptomes incelenmesini sağlayan bir referans genom olmadan de novo monte edilebilir okur. RNA-DİZ teknolojisi aynı zamanda ek yeri izoformları 10-12, yeni transkriptler, 13 ve gen füzyonlarının 14 tespit etmek için kullanılmıştır. Protein kodlayan genlerin saptanması ek olarak, RNA Dizi aynı zamanda gibi kodlayıcı olmayan RNA'lar, transkripsiyonu seviyesini roman tespit ve analiz etmek için kullanılabilir uzun siRNA'nın vb 18 RNA 15,16, mikroRNA 17, kodlayıcı olmayan. Çünkü tBu yöntemin de hassasiyeti, tekli nükleotit varyasyonları 19,20 tespiti için kullanılmıştır.
RNA-Seq teknolojisinin gelişiyle önce, mikrodizin gen ifadesi profili analiz etmek için kullanılan ana yöntem oldu. Önceden tasarlanmış probları sentezlenebilir ve daha sonra, mikro-dizi slayt 21 oluşturmak üzere bir katı yüzeye bağlanır. mRNA, ekstre edilmiş ve cDNA'ya dönüştürülür. Ters transkripsiyon işlemi sırasında, floresanla işaretlenmiş nükleotidlerin cDNA dahil edilir ve cDNA mikrodizi slaytlar üzerine hibridize edilebilir. Belirli bir noktaya toplanan sinyalin yoğunluğu, bu noktada 21 spesifik prob bağlanma cDNA'nın miktarına bağlıdır. RNA-Seq teknolojisi ile karşılaştırıldığında, mikrodizin çeşitli sınırlamalar vardır. RNA-Seq teknoloji kullanımını zaman sınırlar görece yüksek arka plan düzeyinde roman transkript tespit edebiliyor iken Birincisi, mikrodizin, gen şerhin önceden varolan bilgiye dayanır gene ifade seviyesi düşüktür. Bağlı arka plan ve sinyallerin doymaya mikrodizisinin hassasiyeti de, yüksek ve aşağı ifade edilen genlerin 7,22 ile sınırlıdır, oysa Bunun yanı sıra, RNA-Dizi teknoloji, algılama (8.000 kat) 7 çok daha yüksek bir dinamik aralığı vardır. Son olarak, mikrodizi probları bir örnek 23 içindeki farklı transkriptlerinin nispi temsil seviyelerini karşılaştırırken sonuçları daha az güvenilir hale hibritleştirme verimlilikleri, farklıdır. RNA-Seq mikrodizisinden üzerinden pek çok avantajı olmasına rağmen, veri analizi karmaşık. Bu, birçok araştırmacılar hala RNA-Sek yerine mikrodizisini kullanmak nedenlerinden biridir. Çeşitli biyoinformatik araçları RNA-Dizi veri işleme ve analiz 24 için gereklidir.
Birkaç nesil dizileme (NGS) platformlar arasında, 454, Illumina, KATI ve İyon Torrent en yaygın kullanılan olanlardır. 454 ilk ticari NGS platform oldu. Diğer sıralama platformlar aksineBu tür Illumina ve bir katı halinde, 454 platformu okunmayacak oluşturur uzunluğu 25 (ortalama 700 baz kaydeder). Nedeniyle daha uzun daha yüksek verim 25 monte onların için transcriptiome ilk karakterizasyonu için daha iyi okur. 454 platformunun ana dezavantajı dizisinin megabase başına yüksek maliyet. Oluşturmak Illumina ve KATI platformları sayısının artması ve kısa uzunluklarda okur. Dizisinin megabase başına maliyet 454 platformu çok daha düşüktür. Nedeniyle Illumina ve SOLID platformları için okur kısa, çok sayıda, veri analizi çok fazla hesaplama yoğundur. İyon sel platformu için sıralama için alet ve reaktiflerin fiyatı ucuzdur ve sıralama süresi 25 daha kısadır. Ancak, hata oranı ve dizinin megabase başına maliyeti yüksek Illumina ve SOLID platformlara karşılaştırılır. Farklı platformlar kendi avantajları ve dezavantajları vardır ve veri analizi için farklı yöntemler gerektirir. Platform sıralama amacı ve finansman durumuna göre seçilmelidir.
Bu yazıda, bir örnek olarak Illumina RNA-Dizi platform almak. Biz EML hücre kendini yenileme ve farklılaşma anahtar düzenleyicileri araştırmak için bir model sistem olarak EML hücresi kullanılan ve ifade seviyesi hesaplama ve roman transkript tespiti için RNA-Dizi kütüphane yapımı ve veri analizi yöntemleri ayrıntılı sağladı. Biz EML model sisteminde 2 RNA-seq çalışma, zaman fonksiyonel testi (örn ShRNA demonte) hematopoetik farklılaşma erken aşamalarında moleküler mekanizmasının anlaşılması açısından güçlü bir yaklaşım sağlamak ile birleştiğinde bizim önceki yayın göstermiştir, ve bir olarak hizmet verebilir genel olarak hücre kendini yenileme ve farklılaşma analizi için bir model.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. EML Hücre Kültürü ve Sistem ve sıralama Manyetik Hücre kullanma Lin-CD34 + ve Lin-CD34 Hücrelerinin Ayırma Floresan aktive Hücre Ayırma Yöntemi
- Kök hücre faktörü toplanması için bebek hamster böbrek (BHK) hücre kültür ortamının hazırlanması:
- 37 ° C'de bir hücre kültürü inkübatöründe,% 5 CO2 seviyesinde 25 cm2 bir şişede (Tablo 1)% 10 FBS içeren DMEM aracı madde içinde kültür BHK hücreleri.
- Hücreler 80 genişlediğinde, -% 90 birleştiği, 10 ml PBS ile bir kez hücreleri yıkayın. Tek tabaka için% 0.25 tripsin-EDTA çözeltisi 5 ml ilave edilir ve hücreler, müstakil kadar oda sıcaklığında (RT) 1-5 dakika boyunca kuluçkalayın.
- Pipet hafifçe aşağı çözüm yukarı ve hücrelerin öbekler kırmak için. Tripsin aktivitesi durdurmak için şişeye bitmiş DMEM 5 ml. Oda sıcaklığında 5 dakika boyunca 200 x g'de santrifüj ile hücreler toplanır.
- Ortamı çıkarın ve taze BHK hücre kültür ortamı, 10 ml hücre pelletini. Transferi 2 yeni bir 75 cm'lik 2 şişeye aşama 1.1.4 gelen hücre süspansiyonu ilave edildi ve şişeye taze BHK hücre kültür aracı maddesi 48 ml ilave ediniz.
- Kültür BHK iki gün hücreler ve kültür ortamının toplanması. Geçişi, bir 0.45 um filtre içinden ortamı. Bir sonraki kullanıma kadar -20 ° C 'de orta saklayın.
- EML hücre kültürü:
- Bir hücre kültürü inkübatöründe 37 ° C'de,% 5 CO2 seviyesinde BHK hücre kültür ortamı (Tablo 1) ihtiva eden EML bazik bir ortamda kültür EML hücreleri (süspansiyon).
- Tepe yoğunluğu en az 6 x 10 5 hücre / ml düşük hücre yoğunluğunda (0.5-5 x 10 5 hücre / ml) EML hücreleri koruyun. 1 oranında her 2-3 günde bir hücreleri bölün: 5'tir. Passage EML hücreleri nazikçe ve 10 nesiller için pasaj sonra kültürünü atın.
- Soyu pozitif hücrelerin tükenmesi:
- 200 xg FO santrifüj ile EML hücreler hasatR, 5 dakika, bir kez PBS ile hücreleri yıkayın. 5 dakika boyunca 200 x g'de santrifüj ile hücreler toplanır.
- Hücrelerin PBS ile yeniden süspanse edin ve bir hemositometre ile hücreleri sayın. (Hücre izolasyon sisteminin sağlayıcı tarafından sunulan talimatlara bakınız) hücre sayısına bağlı olarak takip eden hücre ayrıştırma aşamasında antikor konsantrasyonunu belirler.
- Soy negatif (Lin-) izole nesilli antikor kokteyli (biyotin-konjuge monoklonal antikor kokteyli hücreleri, CD5, CD45R (B220), CD11b, anti-Gr-1 (Ly-6G / C) 7-4 ve Der-119 ) ve üreticinin talimatlarına göre, bir manyetik etkinleştirilmiş hücre tasnif sistemi.
- Lin-CD34 + ve Lin-CD34 hücre ayrımı:
- 5 dakika boyunca 200 xg'de adım 1.3.3 gelen Lin hücreleri aşağı doğru döndürün. PBS ile hücre pelletini ve bir hemositometre ile hücreleri sayın.
- FACS tamponu ile iki kez hücreler yıkanır ve 200 x g'de, hücreleri topaklamak5 dakika karıştırıldı.
- Sırasıyla sayı 1, 2, 3, 4, 5, beş, 1.5 ml mikrosantrifüj tüpleri etiketleyin. 10 6 hücre (tüp başına 10 6 hücre) başına 100 ul FACS tamponu ile tekrar süspansiyon hücreleri.
- Tüp 1 ve tüp 2 Anti-Mouse CD34 FITC antikor 1 ug ekleyin ve hafifçe tüpleri karıştırın.
- Karanlıkta 1 saat boyunca 4 ° C 'de tüm tüpler inkübe edin.
- Borunun 1, APC-konjuge Köken Kokteyl antikor 0.25 boruyu 3 PE-konjuge anti-Sca1 antikorun ug ve 20 ul ila Köken Kokteyl antikorlar, APC-konjuge PE-konjuge anti-Sca1 antikorun 0.25 ug ve 20 ul ekle tüp 4.
- Nazikçe tüpleri karıştırın ve karanlıkta bir 30 dakika boyunca 4 ° C'de kuluçkalayın.
- Hücrelere FACS tampon 300 ul ilave edin ve 5 dakika boyunca 200 xg'de hücreleri aşağı doğru döndürün.
- Üç kez FACS tampon 500 ul hücreleri yıkayın.
- FACS met 500 ul hücre pelletiniffer.
- Tazminat kurma borular 2, 3, 4, ve 5 'de hücreleri kullanır. FACS Aria kullanarak tüp 1 Lin-SCA + CD34 + ve Lin-SCA-CD34 hücreleri izole.
2. Yüksek verim dizinleri için RNA Hazırlama ve Kütüphanesi İnşaatı
- İzolasyon, kalite analizi ve RNA ölçümü:
- Üretmektedir 'protokol takip edilen Lin-CD34 + ve sırasıyla TRIzol kullanılarak Lin-CD34 hücrelerden toplam RNA ekstrakte edin.
- Üreticinin protokolü takip edilerek, kirlenmiş DNA'sı kullanılarak deoksiribonükleaz I (DNase I) çıkarın. İsteğe bağlı olarak, daha fazla kullanım için bu aşamada -80 ° C'de RNA saklayın.
- Tedarikçi tarafından sunulan talimatlara göre Bioanalyzer kullanan toplam RNA kalitesini değerlendirmek. RNA Bütünlük sayısı (RIN) 9 daha lager RNA örneği kullanın.
- Kütüphane İnşaat ve yüksek verim sıralama:
NOT: Bu protokol Illumina platformu kullanılarak RNA-seg açıklar. IçinDiğer sıralama platformlar, farklı kütüphane hazırlama yöntemleri gereklidir.- Kütüphane hazırlanması için numune başına yüksek kaliteli toplam RNA 0,1-4 mikrogram kullanın. Normal olarak, toplam RNA, 2 ug 10 5 EML hücrelerinden elde edilebilir.
- RNA, saflaştırma ve parçalanma, birinci ve ikinci dizi cDNA sentezi, son tamir, 3, RNA'ya dizileme örnek hazırlama sisteminin kullanımı, 'sağlayıcının talimatlarından ayrıntılı standart prosedürler takip edilerek, PCP, adaptör ligasyonu ve PCR amplifikasyonu sona erer.
- Olumlu oligo-dT manyetik boncuklar kullanılarak PolyA mRNA seçin ve mRNA fragmanı.
- CDNA'nın elde edilmesi ve daha sonra çift şeritli cDNA oluşturmak için cDNA, ikinci dalı sentez etmek için, rastgele primerler kullanılarak ters transkripsiyon gerçekleştirin.
- 3 'çıkıntılar ve 5 doldurun' Kaldır DNA polimeraz tarafından çıkıntılar. Adenilat 3 'birbirlerine bağlanması ile ilgili cDNA fragmanlarının önlemek için sona erer.
- DscDNA her iki ucuna multipleks indeksleme adaptörleri ekleyin. DNA fragmanlarının zenginleştirilmesi için PCR gerçekleştirin.
- Bir spektrofotometre kullanılarak kütüphanenin konsantrasyonu hakkında bilgi edinmek için A260 / A280 ölçün.
- Kütüphane kalitesi değerlendirildi ve Bioanalyzer kullanılarak DNA parçalarının boyut aralığı ölçer.
3. Veri Analizi
Bu bölümde kullanılan yazılım referans için (Tablo 2) bakınız.
- Alt analizi için veri dosya işleme:
- .bcl (Baz çağrı dosyası) dönüştürmek casava yazılımı (Illumina, sürüm 1.8.2) kullanarak dosya .fastq dosya.
- Linux sistemi 'Terminal' ateş. Bir Illumina HiSeq2000 dizileme makineden veri dosyasını içeren veri klasörüne gidin. Sonuç klasör 'NASboy1 / JiaqianLabData / HiSeq_RUN / 2013_07_11 / 130627_SN860_0309_A_2013-166_H0PW9ADXX /' türü olduğunu varsayalımŞekil S1a içinde komuta ve veri klasörünü girin.
- Linux sistemi casava 1.8.2 yükleyin. , Outputfolder 'Hizalanmamış' olduğunu varsayalım dönüştürmek için yapılandırma dosyası hazırlamak için Şekil S1B yılında komutunu kullanın. Sadece bir .fastq dosya her bir numune için oluşturulan sağlamak için --fastq-küme sayımı 0 seçeneğini kullanın. Oluşturulan .fastq dosyası Gz biçimindedir. Alt analizi (Şekil S1B) için açın.
- 'Hizalanmamış' klasörü oluşturulduktan sonra, 'Hizalanmamış' klasöründe (Şekil S1C) gidin.
- Dönüştürme işlemini başlatmak için Şekil S1D yılında komutunu kullanın. '-j' Parametresi kullanılacak cpu sayısını sağlayan.
- Sistem dönüştürme işlemini bitirdikten sonra, 'Hizalanmamış' klasörü (Şekil S1E) kapsamında sonuç klasörüne gidin.
- <Şekil S1F içinde komutunu kullanın/ Strong> Her numune klasörü altında .fastq dosyasının içine .fastq.gz dosyayı açmak için.
- .bcl (Baz çağrı dosyası) dönüştürmek casava yazılımı (Illumina, sürüm 1.8.2) kullanarak dosya .fastq dosya.
- Roman transkript algılamak ve smokin Suite 26 ile ifade düzeyini değerlendirmek:
- RNA-Seq (elde ucsc sürümü MM9, fare referans genomuna okur eşleştirilmiş sonu haritada http://cufflinks.cbcb.umd.edu/igenomes.html kullanır) Tophat yazılımını kullanarak (versiyon 1.3.3) 27 Papyon mapper (sürüm 0.12.7) 28 okuyun. Tophat ifade seviyesi tahmin doğruluğunu geliştirmek için "-hayır-roman-juncs" seçeneği ile verilir.
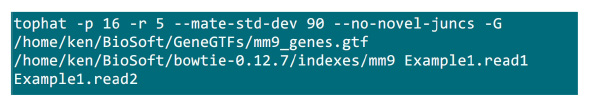
- Haritalama işlemi uygulanacak bir klasörde .fastq dosyaları koydum. Bir paired-end dizileme numune için 2 .fastq dosyaları (Example1.read1, Example1.read2 yeniden adlandırmak) olduğunu varsayalım eşleme (sistem ayarına göre parametrelerini ayarlamak) yapmak için Şekil S2 komutunu kullanın."-p" Parametresi kullanılacak cpu sayısını sağlayan. "R" ve "-mate-std-dev" parametreleri kütüphane QC elde edilen veya (Şekil S2) hizalanmış bir alt kümesi okur varılabilir.
- Kol Düğmesi yazılımını kullanarak RNA transkriptleri okur eşlenen (versiyon 1.3.0) 29 birleştirin. Açıklama bilinen genlerin dosya (tophat tarafından kullanılan aynı .gtf dosyası) ve tophat tarafından üretilen .bam dosyasını kullanarak çalıştırın Kol Düğmeleri.
- Tophat çalışmasını tamamladıktan sonra, aynı klasörde, transcriptome ve tahmin transkript ifade seviyesini oluşturmak için kol düğmeleri çalıştırmak için Şekil S3A yılında komutunu kullanın. 'GenomeSeqMM9' klasöründe 'mm9_repeatMasker.gtf' ve genom dizisi dosyaları UCSC Genom Browser elde edilebilir.
- Elde edilen genes.expr ve transcripts.expr dosyaları genler ve transkript (izoformları) olarak ifade değerini içerir. Kopyalama ve yapıştırmabir Excel dosyası içeriğini dosya ve hesap tablosu uygulaması (Şekil S3 B) ile işleyebilirsiniz.
- Yeni transkript belirlemek için referans 'mm9_genes.gtf' dosyasına çıkan 'transcripts.gtf' dosyası karşılaştırmak için Şekil S3C yılında komutunu kullanın.
- Elde edilen .tmap dosyası karşılaştırma sonucunu içerir. Kopyalayın ve bir Excel dosyası için dosya içeriğini yapıştırın ve elektronik tablo uygulaması ile manipüle. Sınıf kodu ile deşifreleri 'u' (Şekil S3D) sağlanan dosyası .gtf referans ile karşılaştırıldığında 'roman' olarak kabul edilebilir.
NOT: değerleri 0.1 altında ise aşağı analiz kolaylık sağlamak için, 0.1 FPKM değerlerini ayarlayın.
NOT: Adım 3.2.3 - 3.2.6 roman transkriptlerinin 'ifadesi tahminin doğruluğunu geliştirmek isteyenler için isteğe bağlıdır. Haritalama ve transcriptome inşaat r olması gerekir, çünkü bu, çok daha uzun bir zaman alacakun birden çok kez.
- Varsayılan parametrelerini kullanarak Tophat çalıştırın ve daha sonra Şekil S3E içinde komutunu kullanarak oluşturulan .gtf dosyaya kol düğmeleri çalıştırın.
- Şekil S3F yılında komutunu kullanarak referans genom .gtf dosyaya çıkan .gtf dosyasını karşılaştırın.
- Aşama 3.2.2.4 tarif edildiği gibi nihai .tmap dosyasını çözümler. Kopyalayın ve bir Excel dosyası için dosya içeriğini yapıştırın ve elektronik tablo uygulaması ile manipüle. Sınıf kodu ile deşifreleri 'u' olarak kabul edilebilir 'roman' sağlanan dosyası .gtf referans ile karşılaştırıldığında.
- Aşama 3.2.5 sonra, referans .gtf dosyası olarak kullanılabilir klasördeki bir .combined.gtf bir dosya vardır. Yeni transkriptler, daha doğru bir şekilde tahmin FPKM elde etmek için aşama 3.2.1 ve 3.2.2'de tarif edildiği gibi TopHat ve kol düğmeleri ikinci bir çalışma yapılabilir.
- RNA-Seq (elde ucsc sürümü MM9, fare referans genomuna okur eşleştirilmiş sonu haritada http://cufflinks.cbcb.umd.edu/igenomes.html kullanır) Tophat yazılımını kullanarak (versiyon 1.3.3) 27 Papyon mapper (sürüm 0.12.7) 28 okuyun. Tophat ifade seviyesi tahmin doğruluğunu geliştirmek için "-hayır-roman-juncs" seçeneği ile verilir.
- Differentiall Algılamay DESeq paketi 30 kullanılarak genleri dile getirdi.
- DESeq giriş bir ham okuma sayar tablodur. Böyle bir tablo elde etmek için, HTSeq web sitesinden indirilebilir HTSeq Python paketi ile dağıtılan htseq-sayısı komut dosyasını kullanabilirsiniz ( http://www-huber.embl.de/users/anders/HTSeq/doc/count.html ) .
- Samtools, piton ve htseq-sayımı programsare sistemde yüklü olduğundan emin olun. Şekil S4a yılında komutunu kullanarak tophat çıkışından ham okuma sayısı numaralarını edinin.
- 'Raw_Count_Table.txt' Excel kullanarak, 'ExperimentDesign.txt' dosyalarını hazırlayın. Kopyala ve DESeq R paketi (Şekil S4B) için .txt formatında içeriği kaydedebilirsiniz.
- Sistemde R programı yükleyin. Terminalde, yazın 'R' basın ENTER.A ekran mesajı olacak appearas Şekil S4C gösterdi.
- 'Raw_C OkuŞekil S4D yılında komutunu kullanarak R içine ount_Table.txt ',' ExperimentDesign.txt '.
- Şekil S4E yılında komutunu kullanarak DESeq paketi yükleyin.
- R (Şekil S4F) olarak çarpanlara koşulları.
- Normalize sayım masaya olumsuz bimominal testi çalıştırmak için Şekil S4G yılında komutunu kullanın.
- Bir .csv dosyasında çıkış önemli diferansiyel ifade edilen genlerin Şekil S4H yılında komutunu kullanın.
- DESeq giriş bir ham okuma sayar tablodur. Böyle bir tablo elde etmek için, HTSeq web sitesinden indirilebilir HTSeq Python paketi ile dağıtılan htseq-sayısı komut dosyasını kullanabilirsiniz ( http://www-huber.embl.de/users/anders/HTSeq/doc/count.html ) .
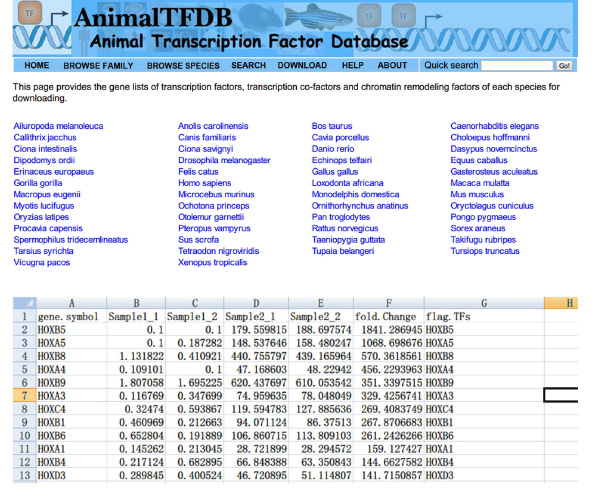
- Excel kullanarak numunelerin genelinde Arama transkripsiyon faktörleri '(TF) FPKM değerleri. Kesişim DE gen masa ve TF'ler tablo. Genler farklı şekilde transkripsiyon faktörlerini ifade hem tabloya ait.
- Web sitesine gitmek http://www.bioguo.org/AnimalTFDB/download.php ve transkripsiyon faktörleri indirin. Ardından <(Excel'de DE transkripsiyon faktörlerini arama strong> Şekil S5).
- UCSC genom tarayıcı görselleştirme için .bigwig dosyası oluşturuluyor.
- Web sitesinden 'bedtools' yazılım paketi indirin https://github.com/arq5x/bedtools2 ve sistemde 31 yazılımı yükleyin. Web sitesinden ucsc aletler 'bedGraphToBigWig' indirin http://hgdownload.cse.ucsc.edu/admin/exe/ ve sistemdeki yazılımı yükleyin.
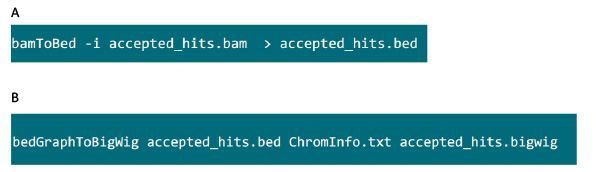
- .bam Dosyasını içeren klasörü, .bed dosyasına tophat tarafından oluşturulan .bam dosyayı dönüştürmek için Şekil S6A yılında komutunu kullanın.
- .Yatakta Dosya üretildikten sonra, .bigwig dosyası oluşturmak için Şekil S6B yılında komutunu kullanın. Dosya 'ChromInfo.txt' Aşağıdaki url elde edilebilir:arget = "_blank"> http://hgdownload.cse.ucsc.edu/goldenPath/mm9/database/chromInfo.txt.gz.
- UCSC Genom Browser bir özel parça gözlemlemek. Web sitesine bakın http://genome.ucsc.edu/goldenPath/help/customTrack.html UCSC genom tarayıcısı kullanarak özel bir iz görüntülemek için nasıl.

Şekil S1: .bcl dosya dönüştürme casava yazılımı kullanarak dosyayı .fastq için.

Şekil S2: Haritalama Tophat kullanarak genom referans okur.
Şekil S3: roman transkript ve ifade seviyesi tahmini Algılama.

Şekil S4: package DESeq kullanarak diferansiyel ifade geni çağrılıyor.

Şekil S5: farklı şekilde ifade transkripsiyon faktörlerinin belirlenmesi.

Şekil S6: veri görselleştirme için haritalama sonucu dönüştürme.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Lin-CD34 + ve Lin-CD34 EML hücrelerinde farklı olarak ifade genlerin analiz etmek için, biz RNA-Dizi teknolojisi kullanılmaktadır. Şekil 1 prosedürlerin iş akışını gösterir. Manyetik hücre sıralama ile soy negatif hücrelerin izolasyonu sonra, Lin-SCA + CD34 + ve FACS Aria kullanılarak Lin-SCA-CD34 hücreleri ayrıldı. Lin-zenginleştirilmiş EML hücreleri, anti-CD34, anti-SCA1 ve soy kokteyl antikor ile boyandı. Sadece Lin- hücreleri Sca1 CD34 ifade analizi için geçitlendi. İki popülasyonları (SCA + CD34 + ve SCA-CD34 EML hücreleri) FACS analizi (Şekil 2) 6 tarafından gözlemlenen olabilir.
Hücrelerin ayrılması sonra, sırasıyla, CD34 + ve CD34-hücrelerinden elde edilen toplam RNA ekstrakte edildi ve RNA kalitesi analiz edilmiştir. RNA Seq verilerinin doğruluğu büyük ölçüde RNA Dizi kütüphane kalitesi üzerine dayanır ve toplam RNA kalitesi, yüksek kaliteli bir kütüphane hazırlanması için hayati önem taşımaktadır. Yüksek kaliteli RNA numunesi 1 arasında bir OD 260/280 değeri olmalıdır.8 ve 2.0. Spektrofotometre kullanılarak, ilave olarak, RNA, kalitesinin daha Bioanalyzer ile daha doğru bir şekilde tayin edilmiştir. 3 9.4'e eşit RIN ile yüksek kalitede bir RNA numunesinin bir sonucunu göstermektedir. RIN değerinden daha büyük 9, sadece, yüksek kaliteli, toplam RNA numunesi, mRNA çıkarma ve daha sonra kütüphane oluşturma işlemleri için kullanıldı.
Ribozomal RNA, hücre içinde RNA en bol türüdür. Şu anda iki temel strateji, rRNA tükenmesi veya poliadenile edilmiş mRNA (poli-A mRNA) pozitif seçimi, kitaplık oluşumu öncesi hedef RNA'nın zenginleştirilmesi için kullanılmıştır. Sigara polyadenylated RNA türlerinin poli-A mRNA seçimi sırasında kaybolur. Buna karşılık, örneğin RiboMinus gibi rRNA tükenmesi yöntemleri olmayan polyadenylated RNA türlerini korumak olabilir. Çalışmamızın amacı, farklı şekilde, böylece biz kütüphane yol yapım önce hedef RNA'lar zenginleştirilmesi için poli-A mRNA seçim yöntemi kullanılır, iki hücre tiplerinde kodlama genleri ifade için bakmaktırction. Kütüphane yapımı tamamlanmış olduğunda, arşivdeki DNA fragmanlarının boyutunu Bioanalyzer kullanılarak dizilim önce kontrol edildi. 4 ila yaklaşık 300 bp parçası boyutu, zirveleri ile kaliteli bir kütüphane göstermektedir.
Sonraki bir adımda, kütüphane, yüksek sekanslama tabi tutuldu. Prensip olarak, artık okumak uzunluk okuma haritalama için yararlı olacaktır. Bu okuma nedeniyle yinelenen genlerin ya da gen aile üyeleri arasında benzerlik birden çok yerde eşleştirilmiş olduğu olasılığını azaltabilir. Çift uç dizileme sekansları parçalarının her iki ucundan olduğu için, seçilen okuma uzunluğu ortalama fragmanları uzunluğunun yarısından daha az olmalıdır. Deneyin temel amacı yerine transkript yapısını inşa ifade seviyesini ölçmek için ise, tek-uç çok fazla bilgi kaybetmeden maliyetini azaltabilir (75 veya 100 bp) okuyun. Paired-end dizileme transkript yapı inşaat ve kısa için daha yararlıdıruzunluğu okuyun maliyetini azaltmak için kullanılabilir. Yeterli fonlama mevcut olduğunda, kesinlikle, uzun okumak uzunluk tercih edilmektedir.
Diferansiyel ifade analizi için, DESeq başka birçok alternatif algoritmaları vardır. 32 cuffdiff isimli kol paketinde yer alan bir de vardır. DESeq en yaygın olarak kullanılan sayısı dayanan DE gen analiz algoritmaları biridir. Negatif binom dağılımı - DESeq yöntem iyi karakterize istatistik modeline dayanmaktadır. Bizim tecrübelerimize göre, DESeq cuffdiff karşılaştırmak daha kararlı. Cuffdiff Erken versiyonları sıklıkla DE genlerin önemli ölçüde farklı numaralarını vermek. Bu nedenle burada DE analizi için DESeq kullanılır.
Transkripsiyon faktörleri hücre kaderinin belirlenmesi için çok önemlidir çünkü, biz önemli ölçüde farklı şekilde ifade transkripsiyon üzerinde duruldu faktör 33. TF'ler Lin-CD34 + ve Lin-CD34 arasında> 1.5 kat bulundu edildi değişti ve Figür (ısı haritasında gösterilmektedire 5) 2. Özellikle, Lin-CD34 + hücrelerinde Tcf7 göreceli ifade seviyesi Lin-CD34 hücrelerinde daha fazla, 100 kat daha fazladır. Böylece Tcf7 EML hücre kendini yenileme ve farklılaşma 2 düzenlenmesinde Tcf7 'ın fonksiyonunu onaylamak için ileri ChIP-Dizilimine (Kromatin Immunoprecipitation ve dizileme) analizi ve fonksiyonel test için seçildi.

Şekil 1:. Prosedürlerin akışı Lin-CD34 + ve Lin-CD34 hücreleri manyetik bir hücre ayırma sistemi ve floresans ile aktive edilmiş hücre tasnifi, ayırma yöntemi ile ayrılır. Toplam RNA, mRNA saflaştırma ve kitaplık oluşumu, ardından ekstre edilmiştir. Kütüphane kalitesi analizi sonra, numuneler, yüksek sekanslama için tabi tutulmuştur. Veri analizi ve diferansiyel olarak transkripsiyon faktörlerini ifade edildi tespit edilmiştir.

Şekil 2:. Lin-CD34 + ve Lin-CD34 EML 6 Lin EML hücreleri manyetik hücre sıralama ile zenginleştirilmiştir hücrelerinin ayrılması. Lin- hücreleri, anti-CD34, anti-SCA1 ve soy kanşım antikor ile boyandı. Lin- hücrelerinin CD34 ve SCA1 ekspresyonu için geçitlendi. Lin-CD34 + SCA + ve Lin-CD34-Ölçeğin EML hücre popülasyonları dizildi.

Şekil 3:., Yüksek kaliteli, toplam RNA numunesinin bir temsili toplam RNA kalitesi, Bioanalyzer ile değerlendirildi. RNA Bütünlük sayısı 9.4 (FU, Floresan Birimleri) 'dir.

Şekil 4:. Eşli-End kütüphane parçaları boyut aralığı Kütüphanenin DNA büyüklüğü dağılımı Bioanalyzer kullanılarak analiz edilmiştir. En fragmanları, 250-500 bp'lik boyut aralığı içinde bulunmaktadır.

Şekil 5:. Farklı biçimde eksprese transkripsiyon faktörleri, Lin-CD34 + hücrelerinin ve Lin-CD34 hücreleri 2 arasında (> 1.5 kat), her hücre türü için, iki bağımsız deney gerçekleştirilmiştir. Kırmızı renk ve aşağı-regüle genler yeşil renk olarak gösterildiği gibi yukarı-regüle genler belirtilmiştir.
Tablo 1: Tampon ve Hücre kültürü ortamları.
| Yazılım | Kullanım | Referans | |||
| 1.2.7 Papyon | Haritalama için tophat tarafından kullanılan | [28] | |||
| Tophat 1.3.3 | Haritalama referans genom geri okur | [27] | |||
| Kol Düğmeleri 1.3.0 | Transkript inşaat ve ifade seviyesi tahmini | [29] | |||
| DESeq 1.16.0 | Diferansiyel ifade analizi | [30] | Bedtools 2.18 | .bed Dosyasının içine .bam dosyasını dönüştürmek | [31] |
| bedGraphToBigWig | Dosyayı .bigwig için .Yatakta dosya dönüştürme | http://genome.ucsc.edu/ |
Tablo 2: veri analizi için yazılım listesi.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Memeli transcriptome 34-38 çok karmaşık. RNA-Seq teknoloji It gen ifadesi analizi için diğer yöntemlere göre birçok avantajı vardır transcriptome analizi, roman transkript tespiti ve tek nükleotid varyasyon keşif vb çalışmalarında önemli bir rol oynamaktadır. Girişte de belirtildiği gibi, mikrodizi hibridizasyon eserler üstesinden gelir ve yeni bir transkript de novo tanımlamak için kullanılabilir. RNA-sıralamasının bir sınırlama Sanger dizileme karşılaştırarak göreli kısa okumak uzunluğudur. Ancak, dizileme teknolojisindeki hızlı gelişme ile, uzunluk sürekli artmaktadır okuyun. Bu yazıda, fare EML hücre kendini yenileme ve farklılaşma potansiyel anahtar regülatörleri belirlemek için bu teknolojiyi kullanarak ayrıntılı yöntemler sağlar.
Bu protokol için ilk adım EML hücre kültürüdür. EML bir hematopoetik öncül hücre hattıdır ve olsa da olabilirSCF ile büyük miktarda yayılır. EML hücre kültürleme durumu normal ölümsüz hücre çizgileri daha fazla dikkat gereklidir. Hücreler beslenen ve hassas çalışması ile düzenli olarak en pasajlandı edilmelidir; aksi takdirde hücreler, kendini yenileme ve farklılaşma özellikleri değiştirmek ve hücre ölümünü geçirebileceği. Hücreleri yeterince topladıktan sonra ilk adım olarak, biz bir manyetik aktif hücre sıralama sistemini kullanarak soy negatif hücreleri izole. Sonra flüoresanslı etkinleştirilmiş hücre tasnif kullanılarak CD34 + ve CD34 hücreleri ayrıldı. EML hücreleri normalde ayrılıktan sonra benzer olmalıdır RNA ekstraksiyon ve CD34 + 'nın numaraları ve CD34 hücreleri için kullanmadan önce en az 10 nesilleri pasajlanır. Iki nüfus hücre sayısında büyük farklılıklar varsa, o kültürü atmak ve kültürü için hücre stokunun başka bir tüp yeniden çözülme önerilir.
CD34 + ve CD34 hücre ayrılmasından sonra, toplam RNA ekstraksiyonu, bu st için önemli bir adım gerçekleştirildiUDY. Yüksek kaliteli RNA dizileme verilerin doğruluğunu vaat yüksek kaliteli kütüphane, yapımı için temel oluşturur. Bu kritik aşama olarak, RNase ile herhangi bir temas kaçınılmalıdır. Tüm reaktifler ücretsiz RNaz edilmelidir. Bu RNA işlerken her zaman eldiven giymek önemlidir. Yüksek kaliteli RNA örnek 1.8 ve 2.0 arasında bir OD 260/280 değeri vardır. RNA içeren sulu faz toplarken, RNA numune ile herhangi bir organik faz taşımak için dikkatli olun. Bu tür RNA fenol ya da kloroform gibi herhangi bir artık organik çözücüler, 1,65 daha düşük bir OD260 / 280 değeri ile sonuçlanacaktır. OD260 / 280 değer 1.65'ten düşükse, etanol ile tekrar RNA hızlandırabilir. % 75 etanol ile yıkandıktan sonra değil, overdry RNA pelet yok. Kurutma RNA topağı tam RNA çözünürlüğünü etkiler ve RNA düşük verime yol açacaktır.
Bu protokol için bir sonraki önemli adım, kütüphane hazırlıktır. Toplam RNA ekstraksiyonu, kirlenmiş DNA'nın uzaklaştırılması için DNase kullanılmasına yönelik bir aşamasından sonra ıDNA kontaminasyonu kullanılan toplam RNA miktarının yanlış tahmininde neden olabilir beri s, önerilir. Bu sonrası uzun süreli depolama beri, RNA izolasyonu hemen sonra aşağı yordamı gerçekleştirmek ve prosedür-çözdürme dondurma tavsiye edilir, RNA bir dereceye kadar düşer. RNA izolasyonu sonra takip eden aşamalar hemen gerçekleştirilemez durumunda, -80 ° C 'de RNA saklayın. Toplam RNA, mRNA saflaştırma ve cDNA sentezi için kullanılmasından önce, kalitesi her zaman kontrol edilmelidir. Sadece yüksek kalitede RNA, bir kütüphane hazırlanması için kullanılabilir. Düşük kaliteli veya bozulmuş RNA kullanarak 'bitiyor fazla temsil 3'ün neden olabilir. Sıralamadan önce, kütüphane kalite maksimum dizilim verimliliği sağlamak değerlendirildi.
Veri analizi bölümünde, bir referans transcriptome olmadan Cufflinks bir çalışma gerçekleştirdikten sonra, biz ikinci kez dosya ve çalıştırmak tophat ve kol düğmeleri .gtf bir referans oluşturulması için bilinen transkript ile yeni transkript birleştirdi.Bu sadece bir kez çalışıyor daha doğru FPKM tahmin temin beri bu iki çalışma prosedürü, tavsiye edilir. Veri analizi sonra, farklı şekilde eksprese edilen genler belirlenmiştir. Alt deneyler in vitro ve in vivo olarak genlerin işlevini doğrulamak için gerçekleştirilebilir. Daha önceki yayın 2, biz önemli ölçüde farklı şekilde ifade transkripsiyon faktörlerini seçti ve kromatin imunopresipitasyon ve sıralama (ChIP-Seq) yaparak bu faktörlerin genom bağlama bölgesini belirledi. Buna ek olarak, Tcf7 fonksiyonel etkisini test etmek için shRNA yok etme deneyi uygulanabilir. Bu aşağı doğru düzenlenir genler önemli ölçüde CD34 + hücrelerinde zenginleştirilmiş bulunmuştur ise Tcf7 demonte hücrelerinde yukarı düzenledi genler yüksek CD34 hücrelerinde zenginleştirilmiş genler bulundu. Bu nedenle, Tcf7 portatif hücrelerin gen ekspresyon profili, bir model sistem olarak EML hücresi kullanılarak, kısmi olarak farklılaşmış CD34 state.Overall doğru kaymışRNA-DNA dizilemesi ve fonksiyonel deneyleri ile birlikte, bunları tespit ve EML hücre kendini yenileme ve farklılaşmasının önemli bir düzenleyicisi olarak Tcf7 doğruladı.
Subscription Required. Please recommend JoVE to your librarian.
Materials
| Name | Company | Catalog Number | Comments |
| Antibiotic-Antimycotic | Invitrogen | 15240-062 | BHK cell culture |
| Anti-Mouse CD34 FITC | eBioscience | 11-0341-81 | FACS sorting |
| Anti-Mouse Ly-6A/E (Sca-1) PE | eBioscience | 12-5981-81 | FACS sorting |
| APC Mouse Lineage Antibody Cocktail | BD Biosciences | 558074 | FACS sorting |
| BD FACSAria Cell Sorter | BD Biosciences | Special offer sysmtem | FACS sorting |
| Corning™ Cell Culture Treated Flasks 75 cm2 | Corning incorporated | 430641 | Cell culture |
| Corning™ Cell Culture Treated Flasks 25 cm2 | Corning incorporated | 430639 | Cell culture |
| Deoxyribonuclease I, Amplification Grade | Invitrogen | 18068-015 | Library preparation |
| DMEM | Invitrogen | 11965-092 | BHK cell culture |
| DPBS | Gibco | 14190 | Cell culture |
| HI FBS | Invitrogen | 16140071 | BHK cell culture |
| Horse Serum | Invitrogen | 16050-122 | EML cell culture |
| IMDM | HyClone | SH30228.02 | EML cell culture |
| L-Glutamine | Invitrogen | 25030-081 | Cell culture |
| Lineage Cell Depletion Kit, mouse | Miltenyi Biotec | 130-090-858 | Isolation of lineage negative cells |
| NanoVue Plus spectrophotometer | GE Healthcare | 28-9569-62 | Quality control |
| Thermo Scientific™ Napco™ 8000 Water-Jacketed CO2 Incubators | Thermo Scientific | 15-497-002 | Cell culture |
| Penicillin-Streptomycin | Invitrogen | 15140-122 | EML cell culture |
| TRIzol® Reagent | Invitrogen | 15596-018 | RNA exraction |
| TruSeq™ RNA Sample Prep Kit v2 -Set B (48 rxn) | Illumina | RS-122-2002 | Library preparation |
| 2100 Electrophoresis Bioanalyzer Instrument | Agilent | G2939AA | Quality control |
| 0.25% Trypsin-EDTA | Gibco | 25200 | Cell culture |
| 0.45 µm Syringe Filters | Nalgene | 190-2545 | Cell culture |
References
- Chambers, S. M., Goodell, M. A. Hematopoietic stem cell aging: wrinkles in stem cell potential. Stem Cell Rev. 3, 201-211 (2007).
- Wu, J. Q., et al. Tcf7 is an important regulator of the switch of self-renewal and differentiation in a multipotential hematopoietic cell line. PLoS genetics. 8, (2012).
- Ye, Z. J., et al. Complex interactions in EML cell stimulation by stem cell factor and IL-3. Proceedings of the National Academy of Sciences of the United States of America. 108, 4882-4887 (2011).
- Tsai, S., Bartelmez, S., Sitnicka, E., Collins, S. Lymphohematopoietic progenitors immortalized by a retroviral vector harboring a dominant-negative retinoic acid receptor can recapitulate lymphoid, myeloid, and erythroid development. Genes Dev. 8, 2831-2841 (1994).
- Weiler, S. R., et al. D3: a gene induced during myeloid cell differentiation of Linlo c-Kit+ Sca-1(+) progenitor cells. Blood. 93, 527-536 (1999).
- Ye, Z. J., Kluger, Y., Lian, Z., Weissman, S. M. Two types of precursor cells in a multipotential hematopoietic cell line. Proc Natl Acad Sci U S A. 102, 18461-18466 (2005).
- Wang, Z., Gerstein, M., Snyder, M. RNA-Seq: a revolutionary tool for transcriptomics. Nature reviews. Genetics. 10, 57-63 (2009).
- Chu, Y., Corey, D. R. RNA sequencing: platform selection, experimental design, and data interpretation. Nucleic acid therapeutics. 22, 271-274 (2012).
- Hornett, E. A., Wheat, C. W. Quantitative RNA-Seq analysis in non-model species: assessing transcriptome assemblies as a scaffold and the utility of evolutionary divergent genomic reference species. BMC genomics. 13, 361 (2012).
- Eswaran, J., et al. RNA sequencing of cancer reveals novel splicing alterations. Scientific reports. 3, 1689 (2013).
- Wang, E. T., et al. Alternative isoform regulation in human tissue transcriptomes. Nature. 456, 470-476 (2008).
- Wu, J. Q., et al. Dynamic transcriptomes during neural differentiation of human embryonic stem cells revealed by short, long, and paired-end sequencing. Proceedings of the National Academy of Sciences of the United States of America. 107, 5254-5259 (2010).
- Loraine, A. E., McCormick, S., Estrada, A., Patel, K., Qin, P. RNA-seq of Arabidopsis pollen uncovers novel transcription and alternative splicing. Plant physiology. 162, 1092-1109 (2013).
- Edgren, H., et al. Identification of fusion genes in breast cancer by paired-end RNA-sequencing. Genome biology. 12, 6 (2011).
- Ilott, N. E., Ponting, C. P. Predicting long non-coding RNAs using RNA sequencing. Methods. 63, 50-59 (2013).
- Sun, L., et al. Prediction of novel long non-coding RNAs based on RNA-Seq data of mouse Klf1 knockout study. BMC bioinformatics. 13, 331 (2012).
- Luo, S. MicroRNA expression analysis using the Illumina microRNA-Seq Platform. Methods in molecular biology. 822, 183-188 (2012).
- Bolduc, F., Hoareau, C., St-Pierre, P., Perreault, J. P. In-depth sequencing of the siRNAs associated with peach latent mosaic viroid infection. BMC molecular biology. 11, 16 (2010).
- Chepelev, I., Wei, G., Tang, Q., Zhao, K. Detection of single nucleotide variations in expressed exons of the human genome using RNA-Seq. Nucleic acids research. 37, 106 (2009).
- Djari, A., et al. Gene-based single nucleotide polymorphism discovery in bovine muscle using next-generation transcriptomic sequencing. BMC genomics. 14, 307 (2013).
- Murphy, D. Gene expression studies using microarrays: principles, problems, and prospects. Advances in physiology education. 26, 256-270 (2002).
- Chen, K., et al. RNA-seq characterization of spinal cord injury transcriptome in acute/subacute phases: a resource for understanding the pathology at the systems level. PLoS one. 8, 72567 (2013).
- Marioni, J. C., Mason, C. E., Mane, S. M., Stephens, M., Gilad, Y. RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome research. 18, 1509-1517 (2008).
- Ramskold, D., Kavak, E., Sandberg, R. How to analyze gene expression using RNA-sequencing data. Methods in molecular biology. 802, 259-274 (2012).
- Glenn, T. C. Field guide to next-generation DNA sequencers. Mol Ecol Resour. 11, 759-769 (2011).
- Trapnell, C., et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nature protocols. 7, 562-578 (2012).
- Trapnell, C., Pachter, L., Salzberg, S. L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 25, 1105-1111 (2009).
- Langmead, B., Trapnell, C., Pop, M., Salzberg, S. L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome biology. 10, 25 (2009).
- Trapnell, C., et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature. 28, 511-515 (2010).
- Anders, S., Huber, W. Differential expression analysis for sequence count data. Genome biology. 11, 106 (2010).
- Quinlan, A. R., Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 26, 841-842 (2010).
- Cheranova, D., et al. RNA-seq analysis of transcriptomes in thrombin-treated and control human pulmonary microvascular endothelial cells. J Vis Exp. , (2013).
- Zhang, H. M., et al. AnimalTFDB: a comprehensive animal transcription factor database. Nucleic acids research. 40, 144-149 (2012).
- Wu, J. Q., et al. Systematic analysis of transcribed loci in ENCODE regions using RACE sequencing reveals extensive transcription in the human genome. Genome Biol. 9, 3 (2008).
- Wu, J. Q., et al. Large-scale RT-PCR recovery of full-length cDNA clones. Biotechniques. 36, 690-696 (2004).
- Wu, J. Q., Shteynberg, D., Arumugam, M., Gibbs, R. A., Brent, M. R. Identification of rat genes by TWINSCAN gene prediction, RT-PCR, and direct sequencing. Genome Res. 14, 665-671 (2004).
- Dewey, C., et al. Accurate identification of novel human genes through simultaneous gene prediction in human, mouse, and rat. Genome Res. 14, 661-664 (2004).
- Wu, J. Characterize Mammalian Transcriptome Complexity. , LAP Lambert Academic Publishing. (2011).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}