

Summary
RNA 시퀀싱과 생물 정보학 분석은 마우스 EMLcells의 린-CD34 + 린-CD34- 부분 집단에서 유의 및 발현 된 전사 인자를 식별하는 데 사용되었다. 이들 전사 인자는 자기 갱신 린-CD34 +를 부분적으로 분화 된 린 - CD34- 세포 사이의 스위치를 결정하는데 중요한 역할을 할 수도있다.
Abstract
조혈 줄기 세포 (조혈 모세포)는 백혈병과 림프종 등 많은 질병에 환자의 조혈 시스템을 다시 이식 치료를 위해 임상 적으로 사용된다. 조혈 모세포의자가 재생과 분화를 조절하는 메커니즘을 해명하는 연구 및 임상 사용을 위해 조혈 모세포 응용 프로그램에 대한 중요하다. 그러나 의한 시험 관내에서 증식 그들의 무능력 조혈 모세포의 대량을 얻을 수 없다. 이 장애물을 극복하기 위해, 우리는 본 연구를위한 모델 시스템으로, 마우스 골수 유래 세포주, EML (적혈구, 골수 및 림프) 세포주를 사용 하였다.
RNA 시퀀싱 (RNA-SEQ)는 점점 유전자 발현 연구를위한 마이크로 어레이를 대체하는 데 사용되어왔다. 여기서는 EML 세포 자기 갱신 및 분화의 조절에 중요한 요소 가능성을 조사하기 위해 RNA-SEQ 기술을 사용하는 상세한 방법을보고한다. 이 문서에서 제공하는 프로토콜은 세 부분으로 나누어 져 있습니다. 첫 번째 파T는 어떻게 문화 EML 세포와 별도의 린-CD34 + 린-CD34- 세포에 대해 설명합니다. 프로토콜의 두 번째 부분은 총 RNA 준비 및 높은 처리량 시퀀싱에 대한 후속 도서관 건설을위한 자세한 절차를 제공합니다. 마지막 부분은 RNA-SEQ 데이터 분석을위한 방법을 설명하고 린-CD34 + 및 CD34- 린 - 세포 간의 발현 된 전사 인자를 식별하는 데이터를 사용하는 방법을 설명합니다. 가장 현저하게 발현 된 전사 인자는 EML 세포 자기 갱신 및 분화를 제어하는 전위 키 레귤레이터 인 것으로 확인되었다. 이 글의 토론 섹션에서, 우리는이 실험의 성공적인 성능을위한 주요 단계를 강조 표시합니다.
요약하면, 본 연구는 EML 세포에자가 재생과 분화 잠재력 레귤레이터를 식별하기 위해 RNA-SEQ 기술을 사용하는 방법을 제공한다. 확인 된 주요 요인은 시험관과 내가 다운 스트림 기능 분석을 실시한다n 개의 생체.
Introduction
조혈 줄기 세포는 성인 골수 틈새에 주로 존재 희귀 혈액 세포이다. 이들은 혈액을 보충하기 위해 필요한 세포 및 면역 시스템 (1)의 제조를위한 책임이있다. 줄기 세포의 일종으로서, 조혈 모세포는자가 재생과 분화를 모두 할 수있다. 조혈 모세포의 운명 결정을 제어하는 메커니즘을 해명,자가 재생 (self-renewal) 또는 분화 중 하나를 향해, 혈액 질환 연구 및 임상 사용 2 조혈 모세포의 조작에 유용한 지침을 제공 할 것입니다. 연구자들이 직면 한 문제는 조혈 모세포가 유지되고 매우 제한적으로 시험 관내에서 확장 될 수 있다는 것이다; 그들의 자손의 대부분은 부분적으로 문화 2에서 차별화된다.
전체 게놈 규모에서 자기 갱신 및 분화 과정을 조절 키 레귤레이터를 식별하기 위해, 우리는 모델 시스템으로서 마우스 원시 조혈 전구 세포 라인 EML을 사용했다. 목세포주는 쥐의 골수 3,4에서 파생된다. 다른 성장 인자가 공급되면, EML 세포는 시험 관내에서 5 적혈구, 골수 및 림프 세포로 분화 할 수있다. 중요한 것은,이 세포주 배양 배지가 multipotentiality 유지 여전히 줄기 세포 인자 (SCF)를 함유하고에서 다량으로 전파 될 수있다. EML 세포는 자기 갱신 린 - SCA + CD34 +의 아 집단으로 분리 부분적 표면 마커 CD34 및 SCA 6에 근거 린 - SCA-CD34- 세포를 구별 할 수있다. 단기 조혈 모세포, SCA + CD34 + 세포와 마찬가지로 자기 갱신 할 수 있습니다. 빠르게 린-SCA + CD34 + 린-SCA-CD34- 세포의 혼합 인구를 재생 및 6 증식을 계속할 수 SCF, 린-SCA + CD34 + 세포로 치료합니다. 두 집단은 형태에 유사하며 C-키트의 mRNA와 단백질 6 비슷한 수준을 가지고있다. 린 - SCA-CD34- 세포는 IL-3 대신 SCF 3을 함유하는 배지에서 증식 할 수있다. UnveilinG 조혈 초기의 발달 전환 세포 및 분자 메커니즘의 더 나은 이해를 제공 할 것입니다 EML 세포 운명 결정의 핵심 규제.
자기 갱신 린 - SCA + CD34 + 및 부분적으로 분화 된 린 - SCA-CD34- 세포 사이의 기본적인 분자 적 차이를 조사하기 위해, 우리는 발현 된 유전자를 확인하기 위해 RNA-SEQ을 사용했다. 전사 인자는 세포의 운명 결정에 결정적으로 우리는 특히, 전사 인자에 초점. RNA-SEQ의 프로필과 게놈 7,8로부터 전사 된 RNA를 정량화 (NGS) 기술 차세대 시퀀싱 기능을 이용하는 최근에 개발 된 방법이다. 간단히, 총 RNA는 폴리 초기 template.The RNA 템플릿은 다음 역전사 효소를 이용하여 cDNA를로 변환으로 선택하고 조각. cDNA 라이브러리 구축 속담 비 저하 된 RNA를 사용하여 전체 길이의 RNA 전 사체를 매핑하기 위해 중요하다. 끝까지 마음을 위해시퀀싱 포즈, 특정 어댑터 서열의 cDNA의 양단에 추가된다. 그리고, 대부분의 경우, cDNA의 분자는 PCR에 의해 증폭되고, 높은 처리량 방법으로 서열 분석.
서열 분석 후, 생성 된이 기준 게놈 전 사체 데이터베이스에 정렬 될 수 읽는다. 수는 기준 유전자지도를 카운트하고,이 정보를 유전자 발현 량을 추정하는데 사용될 수 있음을 읽는다. 또한 비 유기체 모델 9 transcriptomes의 연구를 가능 기준 게놈없이 드 노보를 조립할 수 읽는다. RNA-SEQ 기술은 또한 접합부 동종 10-12, 신규 전 사체 (13)와 유전자 융합체 (14)를 검출하는데 사용되어왔다. 단백질 코딩 유전자의 검출에 더하여, RNA-SEQ도 같은 비 - 코딩의 RNA의 전사 수준을 소설을 검출하고 분석하는 데 사용될 수 긴 siRNA와 등 (18) RNA 15, 16, 마이크로 RNA (17), 비 - 코딩. 때문에 t의이 방법의 정밀도 그는, 그것은 단일 염기 변이 (19, 20)의 검출에 이용되고있다.
RNA-SEQ 기술의 출현 전에 마이크로 어레이 유전자 발현 프로파일을 분석하기 위해 사용되는 주요 방법이었다. 사전 설계된 프로브를 합성하고,이어서 마이크로 어레이 슬라이드 (21)를 형성하도록 고체 표면에 부착된다. mRNA를 추출하여 cDNA를 변환된다. 역전사 과정에서 형광 표지 된 뉴클레오티드의 cDNA에 편입되고하는 cDNA 마이크로 어레이는 슬라이드에 혼성화 될 수있다. 특정 지점에서 수집 된 신호의 세기는 그 스폿 (21) 상에 특이 적 프로브에 결합 된 cDNA의 양에 의존한다. RNA-SEQ 기술에 비해, 마이크로 어레이는 몇 가지 제한 사항이 있습니다. RNA-SEQ 기술의 사용을 제한 할 때 상대적으로 높은 배경 수준에서 신규 전 사체를 검출 할 수있는 동안 첫째, 마이크로 어레이 유전자 주석의 기존 지식에 의존 GENE 발현 수준이 낮습니다. 때문에 배경 및 신호의 포화, 마이크로 어레이의 정확성은 모두 높은 및 낮은 발현 유전자 7,22에 대한 제한, 반면 게다가, RNA-SEQ 기술은 탐지 (8,000 배) 7 훨씬 높은 동적 범위를 가지고있다. 마지막으로, 마이크로 어레이 프로브는 하나의 샘플 (23) 내의 다른 사체의 상대적인 발현 수준을 비교할 때 결과를 덜 신뢰성있는 그들의 혼성화 효율, 다르다. RNA-SEQ은 마이크로 어레이에 비해 많은 장점을 가지고 있지만, 데이터 분석은 복잡하다. 이것은 많은 연구자들이 여전히 RNA-SEQ 대신 마이크로 어레이를 사용하는 이유 중 하나입니다. 다양한 생물 정보학 도구 RNA-SEQ 데이터 처리 및 분석 (24)에 요구된다.
여러 차세대 시퀀싱 (NGS) 플랫폼 중 454, Illumina 사, SOLID 및 이온 런트는 가장 널리 사용되는 것들이다. (454)는 최초의 상용 NGS 플랫폼이었다. 다른 시퀀싱 플랫폼 달리같은 Illumina의 고체로, 454 플랫폼은 더 이상 읽을 생성 길이는 25 (평균 700 기본 읽기). 인해 더 이상 높은 효율 (25)을 조립 자신에 transcriptiome의 초기 특성에 대한 더 나은 읽습니다. 454 플랫폼의 주요 단점은 시퀀스의 megabase 당 높은 비용이다. 생성 일루미나와 SOLID 플랫폼이 증가 숫자와 짧은 길이로 읽습니다. 시퀀스의 megabase 당 비용은 454 플랫폼보다 훨씬 낮다. 때문에 일루미나와 SOLID 플랫폼 읽고 단락의 큰 숫자로, 데이터 분석이 훨씬 더 계산 집약적이다. 이온 토렌트 플랫폼에 대한 시퀀싱 장비 및 시약의 가격은 저렴하고 시퀀싱 시간은 25 짧습니다. 그러나, 에러율 시퀀스 megabase 당 비용이 높은 일루미나 SOLID 플랫폼과 비교된다. 다른 플랫폼은 자신의 장점과 단점을 가지고 데이터 분석을위한 다른 방법이 필요하다. 괞 찮아TForm 클래스는 시퀀싱 목적과 자금의 가용성에 따라 선택해야합니다.
이 논문에서, 우리는 예를 들어 일루미나 RNA-SEQ 플랫폼을. 우리는 EML 세포자가 재생과 분화에 중요한 레귤레이터를 조사하기 위해 모델 시스템으로서 EML 셀을 사용하고, 발현 량의 계산 및 신규 사체 검출 용 RNA-SEQ 라이브러리 구축 및 데이터 분석의 상세한 방법을 제공 하였다. 우리는 EML 모델 시스템 (2)에서 RNA-SEQ 연구 때 기능 테스트 (예 shRNA를 녹다운) 조혈 분화의 초기 단계의 분자 적 메카니즘을 이해하는 강력한 방법을 제공하여 결합 된 우리의 이전의 간행물에 도시 한와 같이 작용할 수 일반적으로 세포의자가 재생과 분화의 분석을위한 모델.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. EML 세포 배양 및 시스템과 정렬 자기 셀 사용 린-CD34 + 린-CD34- 세포의 분리 형광을 활성화 셀 정렬 방법
- 줄기 세포 인자 컬렉션 베이비 햄스터 신장 (BHK) 세포 배양 배지의 제조 :
- 37 ° C 세포 배양 인큐베이터에서 5 % CO 2에서 25cm 2 플라스크 (표 1)에 10 % FBS를 함유하는 DMEM 배지에서 배양 BHK 세포.
- 세포가 80로 성장할 때 - 90 % 합류, PBS 10 ml로 한 번 세포를 씻으십시오. 단층 0.25 % 트립신 EDTA 용액 5 ㎖를 첨가하고, 세포가 분리 될 때까지 실온 (RT)에서 5 분 동안 세포를 배양한다.
- 피펫은 아래로 부드럽게 솔루션 위는 세포의 덩어리를 분쇄한다. 트립신 활동을 중지하기 위해 플라스크에 완전한 DMEM 5 ML을 추가합니다. 실온에서 5 분 동안 200 XG에서 원심 분리하여 세포를 수집합니다.
- 배지를 제거하고 신선한 BHK 세포 배양 배지 10ml에 세포 펠렛을 재현 탁. 전송이 새로운 75cm이 플라스크에 단계 1.1.4에서 세포 현탁액 ㎖의 플라스크에 신선한 BHK 세포 배양 배지 (48)를 가하여.
- 문화 BHK을 이틀 동안 세포는 배양 배지를 수집한다. 통로 0.45 μm의 필터를 통해 매체. 더 사용할 때까지 -20 ° C에서 매체를 저장합니다.
- EML 세포 배양 :
- 세포 배양 인큐베이터에서 37 ° C, 5 % CO 2에서 BHK 세포 배양 배지 (표 1)을 함유 EML 염기성 배지에서 배양 EML 세포 (현탁액).
- 피크 밀도가 6 미만 × 105 세포 / ml의 낮은 세포 밀도 (0.5~5 × 105 세포 / ㎖)에 EML 세포를 유지한다. 1의 비율로 2-3 일마다 셀을 분할 : 5. 항로 EML 세포를 부드럽게하고 10 세대 계대 후 문화를 폐기합니다.
- 리니지 양성 세포의 고갈 :
- 200 XG에 FO에서 원심 분리에 의해 EML 세포를 수확R 5 분 PBS로 한 번 세포를 씻으십시오. 5 분 200 XG에서 원심 분리하여 세포를 수집합니다.
- PBS로 세포를 재현 탁하고 혈구와 세포를 카운트. (세포 격리 시스템의 제공자에 의해 제공되는 지침 참조) 셀의 개수에 따라 셀 후속 분리 단계에서 항체 농도를 결정한다.
- 리니지 음 (선형 적)을 분리 계보 항체 칵테일 (바이오틴 결합 된 단일 클론 항체의 칵테일을 사용하여 세포 CD5, CD45R (B220), CD11b를, 안티 - GR-1 (LY-6G / C), 7-4 및 테르-119 ) 및 제조업체의 지침에 따라 자기 활성화 세포 분류 시스템.
- 린-CD34 + 린-CD34- 세포의 분리 :
- 5 분 동안 200 XG에서 단계 1.3.3에서 선형 적 세포를 스핀 다운. PBS로 세포 펠렛을 재현 탁하고 혈구와 세포를 카운트.
- FACS 버퍼로 두 번 세포를 씻으 200 XG에서 세포 펠렛5 분.
- 각각 숫자 1, 2, 3, 4, 5 다섯 1.5 ml의 마이크로 원심 튜브 레이블. 10 6 세포 (튜브 당 10 6 세포) 당 100 μL FACS 버퍼로 세포를 재현 탁.
- 관 1 관 2 항 - 마우스 CD34 FITC 항체의 1 μg의를 추가하고 부드럽게 튜브를 섞는다.
- 어둠 속에서 1 시간 동안 4 ° C에서 모든 튜브를 품어.
- 관 (1), APC - 복합 리니지 칵테일 항체의 0.25 관 (3)에 PE - 복합 안티 SCA1 항체의 μg의, 20 μL에에 리니지 칵테일 항체 APC - 복합의 PE - 복합 안티 SCA1 항체 0.25 μg의 20 μl를 추가 관 (4).
- 부드럽게 튜브를 혼합하고 어둠 속에서 추가로 30 분 동안 4 ° C에서 세포를 배양한다.
- 세포에 FACS 버퍼의 300 μl를 추가하고 5 분 동안 200 XG에서 세포를 스핀 다운.
- 3 회 FACS 버퍼 500 μL와 세포를 씻으십시오.
- FACS 용 BU 500 μL에서 세포 펠렛을 재현 탁ffer.
- 보상을 설정하기위한 튜브 2, 3, 4 및 5에서 셀을 사용한다. FACS 아리아를 사용하여 관 (1)의 린-SCA + CD34 + 린-SCA-CD34- 세포를 분리.
2. 높은 처리량 시퀀싱을위한 RNA 준비 및 라이브러리 생성
- 격리, 품질 분석 및 RNA의 정량화 :
- 제조 '프로토콜 다음 린-CD34 + 각각 트리 졸을 사용하여 린-CD34- 세포로부터 전체 RNA를 추출합니다.
- 제조의 프로토콜 다음과 같은 오염 된 DNA를 사용하여 데 옥시 리보 뉴 클레아 제 I (DNase의 I)를 제거합니다. 선택적으로, 더 사용하기 위해이 단계에서 -80 ° C에서 RNA를 저장합니다.
- 공급 업체가 제공하는 지침에 따라 Bioanalyzer를 사용하여 총 RNA의 품질을 평가. RNA 무결성 번호 (RIN) 9 이상의 맥주와 RNA 샘플을 사용합니다.
- 라이브러리 생성 및 높은 처리량 시퀀싱 :
참고 :이 프로토콜은 Illumina의 플랫폼을 사용하여 RNA-SEQ에 대해 설명합니다. 에다른 시퀀싱 플랫폼은 다른 라이브러리 제조 방법이 요구된다.- 라이브러리 준비를 위해 샘플 당 높은 품질의 총 RNA의 0.1-4 μg의를 사용합니다. 보통 총 RNA 2 μg의 10 5 EML 세포로부터 추출 될 수있다.
- RNA 정화 및 분열, 제 1 및 제 2 가닥 cDNA 합성, 최종 수리, 3 RNA 시퀀싱 샘플 준비 시스템을 사용 '공급자의 지침에서 상세한 표준 절차에 따라, adenylation, 어댑터 라이 게이션 및 PCR 증폭을 종료한다.
- 긍정적으로 올리고-DT에게 자기 구슬을 사용하여 폴리 -A의 mRNA를 선택하고 mRNA의를 조각.
- 된 cDNA를 수득하고이어서 이중 가닥의 cDNA를 생성하도록하는 cDNA의 두번째 스트랜드를 합성 임의 프라이머를 사용한 역전사를 수행한다.
- 3 '오버행을하고 5 채우기'제거 DNA 중합 효소에 의해 돌출을. 아데 3 '서로 결찰로부터의 cDNA 단편을 방지 종료한다.
- dscDNA의 양단에 다중 색인 어댑터를 추가합니다. DNA 조각의 농축을위한 PCR을 수행합니다.
- 분광 광도계를 사용하여 라이브러리의 농도에 대한 정보를 획득하기 위해 A260 / A280을 측정한다.
- 라이브러리의 품질을 평가하고 Bioanalyzer를 사용하여 DNA 단편의 크기 범위를 측정한다.
3. 데이터 분석
이 부분에 사용되는 소프트웨어의 참고로, (표 2)를 참조하시기 바랍니다.
- 하류 분석을위한 데이터 파일 처리 :
- .bcl (기본 호출 파일)로 변환 CASAVA 소프트웨어 (Illumina 사, 버전 1.8.2)를 사용하여 파일을 .fastq하는 파일.
- 리눅스 시스템의 '터미널'를 발사. 일루미나 HiSeq2000 시퀀싱 시스템에서 데이터 파일이 들어있는 데이터 폴더로 이동합니다. 결과 폴더 'NASboy1 / JiaqianLabData / HiSeq_RUN / 2013_07_11 / 130627_SN860_0309_A_2013-166_H0PW9ADXX /'를 입력한다고 가정 해 봅시다그림 S1A의 명령과의 데이터 폴더를 입력합니다.
- 리눅스 시스템에서 CASAVA 1.8.2을 설치합니다. , outputfolder '는 정렬되지 않은'가정하자 변환하기위한 구성 파일을 준비도 S1B의 명령을 사용한다. 하나의 .fastq 파일이 각 샘플에 대해 생성하기 위해 --fastq 클러스터 카운트 0 옵션을 사용합니다. 생성 된 .fastq 파일은 gz 인 형식으로되어 있습니다. 다운 스트림 분석 (그림 S1B)를 위해 압축을 풉니 다.
- '정렬되지 않은'폴더가 생성 된 후, '정렬되지 않은'폴더 (그림 S1C)로 이동합니다.
- 변환 프로세스를 시작합니다 그림 S1D의 명령을 사용합니다. '-j'매개 변수는 사용되는 CPU 번호를 제공합니다.
- 시스템이 변환 과정을 완료 한 후, '정렬되지 않은'폴더 (그림 S1E)에서 결과 폴더로 이동합니다.
- <그림 S1F의 명령을 사용하여/ strong>을 각각의 샘플 폴더 아래 .fastq 파일로 .fastq.gz 파일의 압축을 해제합니다.
- .bcl (기본 호출 파일)로 변환 CASAVA 소프트웨어 (Illumina 사, 버전 1.8.2)를 사용하여 파일을 .fastq하는 파일.
- 새로운 성적표를 검색 및 턱시도 스위트 룸 (26)를 이용하여 발현 수준을 평가 :
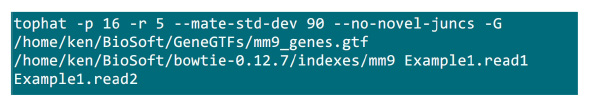
- RNA-SEQ는 (에서 얻은 UCSC 버전 mm9, 마우스 참조 게놈을 읽어 쌍 엔드지도 http://cufflinks.cbcb.umd.edu/igenomes.html 사용) Tophat 소프트웨어를 사용하여 (버전 1.3.3) 27 나비 넥타이는 매퍼 (버전 0.12.7) (28)을 참조하십시오. Tophat는 발현 수준의 추정 정확도를 개선하기 위해 "-no-소설 juncs"옵션과 함께 제공된다.
- 매핑 프로세스가 구현 될 폴더 .fastq 파일을 넣어. , 쌍을 이루는 엔드 시퀀싱 샘플 2 .fastq 파일 (Example1.read1, Example1.read2로 이름을 변경)가 가정의 매핑 (시스템 설정에 따라 매개 변수를 조정)을 할 그림 S2의 명령을 사용합니다."-p"매개 변수는 사용되는 CPU 번호를 제공합니다. "-r"및 "-mate-STD-DEV"파라미터 QC 라이브러리로부터 얻거나 (도 S2)는 정렬의 서브 세트로부터 판독 추론 될 수있다.
- 커프스 소프트웨어를 사용하여 RNA 전 사체에 읽어 매핑 (버전 1.3.0) (29)를 조립합니다. 주석 알려진 유전자의 파일 (Tophat에 의해 사용되는 동일한 .gtf 파일) 및 Tophat에 의해 생산 .bam 파일을 사용하여 실행 커프스.
- Tophat가 실행을 완료 한 후, 같은 폴더에, 사체 및 추정 사체 발현 수준을 구성하는 커프스 단추를 실행하는 그림 S3A의 명령을 사용합니다. 'GenomeSeqMM9'폴더에 'mm9_repeatMasker.gtf'및 게놈 시퀀스 파일은 UCSC 게놈 브라우저에서 얻을 수 있습니다.
- 그 결과 genes.expr과 transcripts.expr 파일은 유전자와 성적 증명서 (동종)의 발현 값이 포함되어 있습니다. 복사 및 붙여 넣기엑셀 파일의 내용은 파일 및 스프레드 시트 응용 프로그램 (그림 S3B)로 조작 할 수 있습니다.
- 신규 한 증명서를 식별하기 위해 참조 'mm9_genes.gtf'파일로 생성 된 'transcripts.gtf'파일을 비교해도 S3C에서 명령을 사용한다.
- 결과 .tmap 파일 비교 결과가 포함되어 있습니다. 복사하여 Excel 파일에 파일의 내용을 붙여 스프레드 시트 응용 프로그램에서 조작 할 수 있습니다. 클래스 코드와 성적 증명서는 'U'는 (그림 S3D) 제공 파일 .gtf 기준에 비해 '소설'로 간주 될 수 있습니다.
참고 : 값이 0.1 미만인 경우 다운 스트림 분석의 편의를 위해, 0.1 FPKM 값을 설정합니다.
참고 : 단계 3.2.3 - 3.2.6은 새로운 성적표 '식 추정의 정확도를 향상하고자하는 사람들을위한 선택 사항입니다. 매핑 및 사체 구조가 연구 될 필요가 있기 때문에, 훨씬 더 긴 시간이 걸릴유엔 번 이상.
- 기본 매개 변수를 사용하여 Tophat 실행 한 다음 그림 S3E의 명령을 사용하여 생성 .gtf 파일에 커프스 단추를 실행합니다.
- 도 S3F에 명령을 사용하여 기준 게놈 .gtf 파일 생성 .gtf 파일 비교.
- 단계 3.2.2.4에 설명 된대로 결과 .tmap 파일을 구문 분석합니다. 복사하여 Excel 파일에 파일의 내용을 붙여 스프레드 시트 응용 프로그램에서 조작 할 수 있습니다. 클래스 코드와 성적 증명서는 'U'는 것으로 간주 할 수있다 '소설'제공 파일 .gtf 기준에 비해.
- 단계 3.2.5 후, 기준 .gtf 파일로서 사용될 수있다 폴더 .combined.gtf 파일이있다. 신규 성적 FPKM보다 정확한 추정을 획득하기 위해 단계 3.2.1 및 3.2.2에 기재된 바와 같이 Tophat과 커프스의 두 번째 실행이 수행 될 수있다.
- RNA-SEQ는 (에서 얻은 UCSC 버전 mm9, 마우스 참조 게놈을 읽어 쌍 엔드지도 http://cufflinks.cbcb.umd.edu/igenomes.html 사용) Tophat 소프트웨어를 사용하여 (버전 1.3.3) 27 나비 넥타이는 매퍼 (버전 0.12.7) (28)을 참조하십시오. Tophat는 발현 수준의 추정 정확도를 개선하기 위해 "-no-소설 juncs"옵션과 함께 제공된다.
- differentiall 감지Y는 DESeq 패키지 (30)를 이용하여 유전자를 표명했다.
- DESeq의 입력은 판독 원시 카운트 테이블이다. 이러한 테이블을 얻으려면, HTSeq 웹 사이트에서 다운로드 할 수 있습니다 HTSeq 파이썬 패키지와 함께 배포되는 htseq 카운트 스크립트를 사용 ( http://www-huber.embl.de/users/anders/HTSeq/doc/count.html ) .
- samtools, Python 및 htseq 카운트 programsare이 시스템에 설치되어 있는지 확인합니다. 그림 S4A의 명령을 사용하여 tophat 출력 원시 읽기 카운트 번호를 얻습니다.
- 'Raw_Count_Table.txt'Excel을 사용하여, 'ExperimentDesign.txt'파일을 준비합니다. 복사 및 DESeq의 R 패키지 (그림 S4B)에 대한이 .txt 형식으로 내용을 저장합니다.
- 시스템에 R 프로그램을 설치합니다. 터미널에 입력 'R'키를 누릅니다 ENTER.A 화면 메시지 것 appearas는 그림 S4C에 보여 주었다.
- 'Raw_C 읽기그림 S4D의 명령을 사용하여 R에 ount_Table.txt ','ExperimentDesign.txt '.
- 그림 S4E의 명령을 사용하여 DESeq 패키지를로드합니다.
- R (그림 S4F)에서 인수 분해 조건.
- 정규화 카운트 테이블에 부정적인 이항식의 테스트를 실행하기 위해 그림 S4G의 명령을 사용합니다.
- .csv 파일에서 출력 중요한 차등 발현 유전자에 그림 S4H의 명령을 사용합니다.
- DESeq의 입력은 판독 원시 카운트 테이블이다. 이러한 테이블을 얻으려면, HTSeq 웹 사이트에서 다운로드 할 수 있습니다 HTSeq 파이썬 패키지와 함께 배포되는 htseq 카운트 스크립트를 사용 ( http://www-huber.embl.de/users/anders/HTSeq/doc/count.html ) .
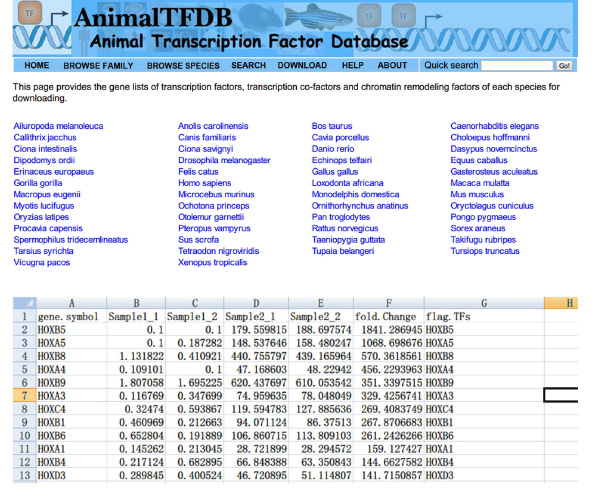
- Excel을 사용하여 샘플에서 조회 전사 인자 '(TF가) FPKM 값. 교차 DE 유전자 테이블과 TF가 테이블. 유전자는 차동 전사 인자를 발현되는 두 테이블에 속한다.
- 웹 사이트로 이동 http://www.bioguo.org/AnimalTFDB/download.php 및 전사 인자를 다운로드합니다. 그런 다음 <(Excel에서 드 전사 인자를 조회 강한> 그림 S5).
- UCSC 게놈 브라우저 시각화를위한 .bigwig 파일을 생성.
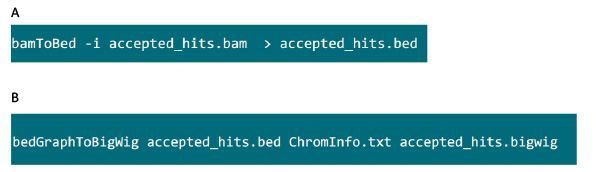
- 웹 사이트에서 'bedtools'소프트웨어 패키지를 다운로드 https://github.com/arq5x/bedtools2 시스템 (31)에 소프트웨어를 설치합니다. 웹 사이트에서 UCSC 도구 'bedGraphToBigWig'를 다운로드 http://hgdownload.cse.ucsc.edu/admin/exe/ 시스템에 소프트웨어를 설치합니다.
- .bam 파일이 들어있는 폴더에서 .bed 파일에 tophat에 의해 생성 .bam 파일을 변환 그림 S6A의 명령을 사용합니다.
- .bed 파일이 생성되면, .bigwig 파일을 생성하기 위해도 S6B에서 명령을 사용한다. 파일 'ChromInfo.txt'는 다음 URL에서 얻을 수 있습니다 :arget = "_blank"> http://hgdownload.cse.ucsc.edu/goldenPath/mm9/database/chromInfo.txt.gz.
- UCSC 게놈 브라우저에 대한 사용자 정의 트랙을 준수하십시오. 웹 사이트를 참조하십시오 http://genome.ucsc.edu/goldenPath/help/customTrack.html UCSC 게놈 브라우저를 사용하여 사용자 정의 트랙을 표시하는 방법에.

그림 S1 : .bcl 파일을 변환하는 CASAVA 소프트웨어를 사용하여 파일을 .fastq합니다.

그림 S2는 : 매핑 Tophat를 사용하여 게놈을 참조하는 읽습니다.
도 S3 : 신규 사체와 발현 량 추정의 검출.

그림 S4 : 패키지를 DESeq 사용하여 차등 발현 유전자를 호출.

도 S5 : 발현 된 전사 인자의 식별.

그림 S6 : 데이터 시각화에 대한 매핑 결과를 변환.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
린 - CD34 + 및 CD34- EML 린 - 세포에서 차별적으로 발현 된 유전자를 분석하기 위해, 우리는 RNA-SEQ 기술을 사용 하였다.도 1은 절차의 흐름을 도시한다. 자기 세포 분류에 의한 혈통 부정적인 세포의 분리 후, 우리는 린-SCA + CD34 + 및 FACS 아리아를 사용하여 린-SCA-CD34- 세포를 분리. 린 풍부한 EML 세포는 항 CD34, 안티 SCA1 리니지 칵테일 항체로 염색 하였다. 만 선형 적 세포 SCA1 및 CD34 발현의 분석을 위해 문이되었다. 두 집단 (SCA + CD34 + 및 SCA-CD34- EML 세포) FACS 분석 (그림 2) (6)에 의해 관찰 될 수있다.
세포 분리 후, 우리는 각각 CD34 + 및 CD34- 세포에서 총 RNA를 추출 RNA의 품질을 분석 하였다. RNA-SEQ 데이터의 정확도는 크게 RNA-SEQ 라이브러리의 질에 의존하고 총 RNA의 품질은 고품질의 라이브러리의 제조에 매우 중요하다. 고품질의 RNA 샘플은 1 사이의 OD 280 분의 260의 값을 가져야한다.8 2.0. 분광 광도계를 사용하는 것 외에도, RNA의 질이 더욱 Bioanalyzer 의해 더 정확하게 평가 하였다. (3)이 9.4과 동일 RIN과 고품질 RNA 샘플의 결과를 보여준다. RIN 값보다 큰 9 만 고품질의 전체 RNA 샘플은 mRNA의 추출 및 후속 라이브러리 구축 절차를 사용 하였다.
리보좀 RNA는 세포에서 RNA의 가장 풍부한 형태이다. 현재 두 가지 전략, rRNA 유전자의 고갈 또는 폴리아 데 닐화의 mRNA (폴리-A의 mRNA)의 긍정적 인 선택은, 라이브러리 생성하기 전에 대상 RNA의 농축에 사용됩니다. 비 폴리아 데 닐화 RNA 종은 폴리 mRNA를 선택하는 동안 손실됩니다. 대조적으로, 예컨대 RiboMinus 같은 rRNA의 고갈 방법 비 폴리아 데 닐화 RNA 종을 보존 할 수있다. 본 연구의 목적은 차등 따라서 우리는 도서관의 constru 전에 대상의 RNA의 농축을위한 폴리-A mRNA의 선택 방법을 사용, 두 종류의 세포로 코딩하는 유전자를 발현을 찾는 것입니다ction의. 라이브러리 구조가 완성 된 때, 라이브러리의 DNA 단편의 크기를 이용하여 시퀀싱 Bioanalyzer 전에 확인 하였다. (4)에서 약 300 bp의 단편 크기 봉우리 양질 라이브러리를 보여준다.
후속 단계에서, 상기 라이브러리는 높은 처리량 시퀀싱 하였다. 원칙적으로, 더 이상 읽을 길이는 읽기 매핑에 도움이 될 것입니다. 이는 판독 인해 중복 된 유전자 또는 유전자 패밀리 구성원 간의 유사성 여러 위치에 매핑 될 확률을 줄일 수있다. 한 쌍의 엔드 시퀀싱 서열 단편의 양단으로부터 아르 같이, 선택된 판독 길이는 평균 단편 길이의 절반 이하이어야한다. 실험의 주요 목표 대신 성적 구조 구축의 발현 수준을 측정하는 경우, 단일 엔드가 너무 많은 정보를 손실하지 않고 비용을 줄일 수있다 (75 또는 100 염기쌍)를 읽었다. 짝 엔드 시퀀싱 성적 증명서 구조의 건설 및 짧은 더 유용합니다길이를 판독하는 비용을 줄일 수있다. 충분한 자금을 사용할 수있을 때 확실히, 더 이상 읽을 길이가 바람직하다.
차등 발현 분석을 위해, DESeq 이외의 많은 다른 알고리즘이있다. 32 cuffdiff라는 커프스 패키지에 포함 된 하나가있다. DESeq는 가장 널리 사용 횟수 기반 DE 유전자 분석 알고리즘의 하나이다. 음 이항 분포 - DESeq 방법은 잘 특성화 된 통계 모델에 기초한다. 우리의 경험에 의하면, DESeq는 cuffdiff에 비해 더 안정적이다. cuffdiff의 초기 버전은 종종 DE 유전자의 유의 한 차이가 번호를 제공합니다. 그러므로 우리는 여기 DE 분석 DESeq을 사용했다.
전사 인자는 세포의 운명 결정에 매우 중요하기 때문에, 우리는 크게 발현 된 전사에 초점을 맞춘 33 요인. TF가 린-CD34 + 린-CD34- 사이> 1.5 배 발견을 변경하고 Figur (히트 맵에 표시됩니다예 5) (2). 특히, 린 - CD34 + 세포가 Tcf7의 상대적 발현 량은 린 - CD34- 세포에 비해 100 배 이상 높다. 따라서 Tcf7는 EML 세포의자가 재생 (self-renewal)과 분화 (2)의 규정에 Tcf7의 기능을 확인하기 위해 추가로 칩 시퀀싱 (염색질 면역 침전 및 염기 서열) 분석 및 기능 테스트를 위해 선택되었다.

그림 1. 절차 플로 린 - CD34 + 및 CD34- 세포 린 - 자성 세포 분리 시스템 및 형광 - 활성화 세포 분류 방법에 의해 분리 하였다. 총 RNA는 mRNA의 정제 및 라이브러리 구축 하였다 추출 하였다. 라이브러리 품질 분석 한 후, 샘플은 높은 처리량 시퀀싱을 하였다. 데이터 분석 및 차등 전사 인자를 발현시켰다 확인되었다.

그림 2 :. 린-CD34 + 린-CD34- EML 6 선형 적 EML 세포가 자기 세포 분류에 의해 농축 된 세포의 분리. 선형 적 세포는 항 CD34, 안티 SCA1 및 계보 혼합 항체로 염색 하였다. 선형 적 세포는 CD34과 SCA1의 표현 문이되었다. 린-CD34 + SCA + 린-CD34-SCA- EML의 세포 집단이 분류되었다.

도 3 :. 고품질의 전체 RNA 시료의 대표가 총 RNA의 품질 Bioanalyzer 의해 평가 하였다. RNA 무결성 번호는 9.4 (FU, 형광 단위)입니다.

그림 4 :. 짝 엔드 라이브러리의 파편 크기 범위 DNA 라이브러리의 크기 분포는 Bioanalyzer를 사용하여 분석 하였다. 대부분의 단편 250-500 bp의 크기 범위 내에있다.

그림 5 :. 발현 된 전사 인자 린-CD34 + 세포와 린-CD34- 세포 2 사이 (> 1.5 배)은 각 세포 유형의 경우, 두 개의 독립적 인 실험을 수행 하였다. 붉은 색과 아래로 조절 유전자가 녹색으로 표시 될 때까지 조절 유전자가 표시됩니다.
표 1 : 버퍼 및 세포 배양 매체.
| 소프트웨어 | 용법 | 참고 | |||
| 1.2.7를 나비 넥타이 | 매핑 Tophat에서 사용 | [28] | |||
| Tophat 1.3.3 | 매핑 참조 게놈을 읽습니다 | [27] | |||
| 커프스 1.3.0 | 성적 증명서의 건설 및 발현 수준 추정 | [29] | |||
| DESeq 1.16.0 | 차등 발현 분석 | [30] | Bedtools 2.18 | .bed 파일로 .bam 파일을 변환 | [31] |
| bedGraphToBigWig | 파일을 .bigwig하는 .bed 파일을 변환 | http://genome.ucsc.edu/ |
표 2 : 데이터 분석을위한 소프트웨어의 목록.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
포유 동물의 사체는 34-38 매우 복잡하다. RNA-SEQ 기술은 유전자 발현 분석을위한 다른 방법에 비해 많은 장점을 가지고 사체 분석, 신규 성적 검출 및 단일 뉴클레오타이드 변이 검색 등의 연구에서 점점 더 중요한 역할을한다. 도입부에서 언급 한 바와 같이, 그 마이크로 어레이 혼성화 아티팩트를 극복하고 새로운 사체 드 노보를 식별하는데 사용될 수있다. RNA 시퀀싱의 한 가지 제한은 생어 시퀀싱에 비해 상대적으로 짧은 읽기 길이입니다. 그러나, 시퀀싱 기술의 급속한 개선으로, 길이가 지속적으로 증가하고 읽습니다. 이 논문에서, 우리는 마우스 EML 세포자가 재생 (self-renewal)과 분화에 잠재적 인 키 레귤레이터를 식별하기 위해이 기술을 사용하는 자세한 방법을 제공합니다.
이 프로토콜의 첫 단계는 키 EML 세포 배양이다. EML는 조혈 전구 세포 라인이며 비록 그것이있을 수SCF와 대량으로 전파. EML 세포의 배양 조건은 일반적인 불멸화 세포주보다 많은주의를 필요로한다. 세포를 공급하고 부드러운 동작에 정기적으로 계대한다; 그렇지 않으면 세포는자가 재생 및 분화 그 특성의 변화 및 세포 사멸을받을 수 있습니다. 충분한 세포를 수집 한 후 첫 번째 단계로, 우리는 자기 활성화 세포 분류 시스템을 사용하여 리니지 음성 세포를 격리합니다. 그런 다음 우리는 형광 활성화 세포 분류를 사용하여 CD34 + 및 CD34- 세포를 분리. EML 세포는 정상적으로 분리 후 유사해야 RNA 추출 및 CD34 +의 번호 및 CD34- 세포를 위해 사용하기 전에 10 미만 세대 계대. 두 집단은 세포 수를 크게 다를 경우, 문화를 버리고 문화에 대한 세포 주식의 또 다른 튜브를 다시 해동하는 것이 좋습니다.
CD34의 + 및 CD34- 세포를 분리 한 후, 총 RNA의 추출은,이 세인트위한 또 다른 중요한 단계를 수행 하였다udy. 고품질 RNA 시퀀싱 데이터의 정확성을 약속 고품질 라이브러리, 건설 용 기재이다. 이 중요한 단계에서의 RNase와 접촉은 피해야한다. 모든 시약은 무료의 RNase해야합니다. 이 RNA를 처리하는 동안 항상 장갑을 착용하는 것이 중요합니다. 고품질의 RNA 샘플은 1.8과 2.0 사이의 OD 280 분의 260 값을가집니다. RNA를 함유하는 수성 상을 수집 할 때, RNA 샘플과 유기 단계를 수행하지 않도록주의하십시오. 이러한 RNA 페놀 또는 클로로포름 등의 잔류 유기 용매는 1.65 미만 OD260 / 280의 값을 초래할 것이다. OD260 / 280의 값이 1.65보다 작 으면, 에탄올로 다시 RNA를 침전. 75 % 에탄올로 세정 한 후,하지 overdry RNA 펠렛을한다. 건조 RNA 펠렛을 완전히 RNA의 용해도에 영향을 RNA의 낮은 수율로 이어질 것이다.
이 프로토콜의 다음 주요 단계는 라이브러리 준비입니다. 총 RNA 추출, DNA의 오염 제거의 DNase를 사용하는 단계 이후 나는DNA 오염 사용한 총 RNA의 양을 잘못 추정 될 수 있으므로들 적극 추천. 그것은 후 장기 저장하기 때문에, RNA 분리 후 즉시 하류 절차를 수행하고 절차 해동 동결하는 것이 좋습니다, RNA는 어느 정도 저하됩니다. RNA 분리 후의 후속 단계가 바로 수행 할 수없는 경우, -80 ° C에서 RNA를 저장한다. 총 RNA는 mRNA의 정제 및 cDNA 합성에 사용되기 전에, 품질이 항상 점검해야한다. 전용 고품질 RNA는 라이브러리 제조를 위해 사용될 수있다. 낮은 품질 또는 성능이 저하 된 RNA를 사용하면 '종료 과잉 표현 (3)으로 이어질 수 있습니다. 시퀀싱 전에 라이브러리 품질 최대 시퀀싱 효율을 보장하기 위해 평가 하였다.
데이터 분석 부에서 기준 사체없이 커프스의 실행을 수행 한 후, 우리는 두 번째 시간 파일 및 실행 Tophat와 커프스 .gtf 기준을 형성하기 위해 공지 된 전 사체와 신규 성적 결합.이 한 번만 실행보다 더 정확한 FPKM 추정을 제공하기 때문에이 두 실행 절차는 좋습니다. 데이터를 분석 한 결과, 발현 된 유전자를 확인 하였다. 하류 실험 관내 및 생체 내에서 유전자의 기능을 확인하기 위해 수행 될 수있다. 우리의 이전이 공보에서는 크게 발현 된 전사 인자를 선택하고 염색질 면역 및 시퀀싱 (서열 번호 칩)을 수행하여 이러한 요소 게놈 결합 부위를 확인 하였다. 또, Tcf7의 작용 효과를 테스트하기 위해 최저 shRNA를 분석 적용. 우리는 아래로 조절 유전자가 크게 CD34 + 세포가 풍부한 것으로 밝혀졌다 동안 Tcf7 넉다운 세포에서, 상향 - 조절 유전자가 매우 CD34- 세포에 농축 유전자는 것을 발견했다. 따라서 Tcf7 넉다운 세포의 유전자 발현 프로파일 모델 시스템으로서 EML 셀을 이용하여, 부분적으로 분화 된 CD34- state.Overall 측으로 시프트RNA 시퀀싱 기술 및 기능 분석과 함께, 우리는 식별되고 EML 세포 자기 갱신 및 분화의 중요한 조절 제로 Tcf7 확인.
Subscription Required. Please recommend JoVE to your librarian.
Materials
| Name | Company | Catalog Number | Comments |
| Antibiotic-Antimycotic | Invitrogen | 15240-062 | BHK cell culture |
| Anti-Mouse CD34 FITC | eBioscience | 11-0341-81 | FACS sorting |
| Anti-Mouse Ly-6A/E (Sca-1) PE | eBioscience | 12-5981-81 | FACS sorting |
| APC Mouse Lineage Antibody Cocktail | BD Biosciences | 558074 | FACS sorting |
| BD FACSAria Cell Sorter | BD Biosciences | Special offer sysmtem | FACS sorting |
| Corning™ Cell Culture Treated Flasks 75 cm2 | Corning incorporated | 430641 | Cell culture |
| Corning™ Cell Culture Treated Flasks 25 cm2 | Corning incorporated | 430639 | Cell culture |
| Deoxyribonuclease I, Amplification Grade | Invitrogen | 18068-015 | Library preparation |
| DMEM | Invitrogen | 11965-092 | BHK cell culture |
| DPBS | Gibco | 14190 | Cell culture |
| HI FBS | Invitrogen | 16140071 | BHK cell culture |
| Horse Serum | Invitrogen | 16050-122 | EML cell culture |
| IMDM | HyClone | SH30228.02 | EML cell culture |
| L-Glutamine | Invitrogen | 25030-081 | Cell culture |
| Lineage Cell Depletion Kit, mouse | Miltenyi Biotec | 130-090-858 | Isolation of lineage negative cells |
| NanoVue Plus spectrophotometer | GE Healthcare | 28-9569-62 | Quality control |
| Thermo Scientific™ Napco™ 8000 Water-Jacketed CO2 Incubators | Thermo Scientific | 15-497-002 | Cell culture |
| Penicillin-Streptomycin | Invitrogen | 15140-122 | EML cell culture |
| TRIzol® Reagent | Invitrogen | 15596-018 | RNA exraction |
| TruSeq™ RNA Sample Prep Kit v2 -Set B (48 rxn) | Illumina | RS-122-2002 | Library preparation |
| 2100 Electrophoresis Bioanalyzer Instrument | Agilent | G2939AA | Quality control |
| 0.25% Trypsin-EDTA | Gibco | 25200 | Cell culture |
| 0.45 µm Syringe Filters | Nalgene | 190-2545 | Cell culture |
References
- Chambers, S. M., Goodell, M. A. Hematopoietic stem cell aging: wrinkles in stem cell potential. Stem Cell Rev. 3, 201-211 (2007).
- Wu, J. Q., et al. Tcf7 is an important regulator of the switch of self-renewal and differentiation in a multipotential hematopoietic cell line. PLoS genetics. 8, (2012).
- Ye, Z. J., et al. Complex interactions in EML cell stimulation by stem cell factor and IL-3. Proceedings of the National Academy of Sciences of the United States of America. 108, 4882-4887 (2011).
- Tsai, S., Bartelmez, S., Sitnicka, E., Collins, S. Lymphohematopoietic progenitors immortalized by a retroviral vector harboring a dominant-negative retinoic acid receptor can recapitulate lymphoid, myeloid, and erythroid development. Genes Dev. 8, 2831-2841 (1994).
- Weiler, S. R., et al. D3: a gene induced during myeloid cell differentiation of Linlo c-Kit+ Sca-1(+) progenitor cells. Blood. 93, 527-536 (1999).
- Ye, Z. J., Kluger, Y., Lian, Z., Weissman, S. M. Two types of precursor cells in a multipotential hematopoietic cell line. Proc Natl Acad Sci U S A. 102, 18461-18466 (2005).
- Wang, Z., Gerstein, M., Snyder, M. RNA-Seq: a revolutionary tool for transcriptomics. Nature reviews. Genetics. 10, 57-63 (2009).
- Chu, Y., Corey, D. R. RNA sequencing: platform selection, experimental design, and data interpretation. Nucleic acid therapeutics. 22, 271-274 (2012).
- Hornett, E. A., Wheat, C. W. Quantitative RNA-Seq analysis in non-model species: assessing transcriptome assemblies as a scaffold and the utility of evolutionary divergent genomic reference species. BMC genomics. 13, 361 (2012).
- Eswaran, J., et al. RNA sequencing of cancer reveals novel splicing alterations. Scientific reports. 3, 1689 (2013).
- Wang, E. T., et al. Alternative isoform regulation in human tissue transcriptomes. Nature. 456, 470-476 (2008).
- Wu, J. Q., et al. Dynamic transcriptomes during neural differentiation of human embryonic stem cells revealed by short, long, and paired-end sequencing. Proceedings of the National Academy of Sciences of the United States of America. 107, 5254-5259 (2010).
- Loraine, A. E., McCormick, S., Estrada, A., Patel, K., Qin, P. RNA-seq of Arabidopsis pollen uncovers novel transcription and alternative splicing. Plant physiology. 162, 1092-1109 (2013).
- Edgren, H., et al. Identification of fusion genes in breast cancer by paired-end RNA-sequencing. Genome biology. 12, 6 (2011).
- Ilott, N. E., Ponting, C. P. Predicting long non-coding RNAs using RNA sequencing. Methods. 63, 50-59 (2013).
- Sun, L., et al. Prediction of novel long non-coding RNAs based on RNA-Seq data of mouse Klf1 knockout study. BMC bioinformatics. 13, 331 (2012).
- Luo, S. MicroRNA expression analysis using the Illumina microRNA-Seq Platform. Methods in molecular biology. 822, 183-188 (2012).
- Bolduc, F., Hoareau, C., St-Pierre, P., Perreault, J. P. In-depth sequencing of the siRNAs associated with peach latent mosaic viroid infection. BMC molecular biology. 11, 16 (2010).
- Chepelev, I., Wei, G., Tang, Q., Zhao, K. Detection of single nucleotide variations in expressed exons of the human genome using RNA-Seq. Nucleic acids research. 37, 106 (2009).
- Djari, A., et al. Gene-based single nucleotide polymorphism discovery in bovine muscle using next-generation transcriptomic sequencing. BMC genomics. 14, 307 (2013).
- Murphy, D. Gene expression studies using microarrays: principles, problems, and prospects. Advances in physiology education. 26, 256-270 (2002).
- Chen, K., et al. RNA-seq characterization of spinal cord injury transcriptome in acute/subacute phases: a resource for understanding the pathology at the systems level. PLoS one. 8, 72567 (2013).
- Marioni, J. C., Mason, C. E., Mane, S. M., Stephens, M., Gilad, Y. RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome research. 18, 1509-1517 (2008).
- Ramskold, D., Kavak, E., Sandberg, R. How to analyze gene expression using RNA-sequencing data. Methods in molecular biology. 802, 259-274 (2012).
- Glenn, T. C. Field guide to next-generation DNA sequencers. Mol Ecol Resour. 11, 759-769 (2011).
- Trapnell, C., et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nature protocols. 7, 562-578 (2012).
- Trapnell, C., Pachter, L., Salzberg, S. L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 25, 1105-1111 (2009).
- Langmead, B., Trapnell, C., Pop, M., Salzberg, S. L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome biology. 10, 25 (2009).
- Trapnell, C., et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature. 28, 511-515 (2010).
- Anders, S., Huber, W. Differential expression analysis for sequence count data. Genome biology. 11, 106 (2010).
- Quinlan, A. R., Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 26, 841-842 (2010).
- Cheranova, D., et al. RNA-seq analysis of transcriptomes in thrombin-treated and control human pulmonary microvascular endothelial cells. J Vis Exp. , (2013).
- Zhang, H. M., et al. AnimalTFDB: a comprehensive animal transcription factor database. Nucleic acids research. 40, 144-149 (2012).
- Wu, J. Q., et al. Systematic analysis of transcribed loci in ENCODE regions using RACE sequencing reveals extensive transcription in the human genome. Genome Biol. 9, 3 (2008).
- Wu, J. Q., et al. Large-scale RT-PCR recovery of full-length cDNA clones. Biotechniques. 36, 690-696 (2004).
- Wu, J. Q., Shteynberg, D., Arumugam, M., Gibbs, R. A., Brent, M. R. Identification of rat genes by TWINSCAN gene prediction, RT-PCR, and direct sequencing. Genome Res. 14, 665-671 (2004).
- Dewey, C., et al. Accurate identification of novel human genes through simultaneous gene prediction in human, mouse, and rat. Genome Res. 14, 661-664 (2004).
- Wu, J. Characterize Mammalian Transcriptome Complexity. , LAP Lambert Academic Publishing. (2011).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}