

Summary
RNA-de sequenciamento e bioinformática análises foram utilizados para identificar fatores de transcrição significativamente e diferencialmente expressos em subpopulações Lin-CD34 + e Lin-CD34 @ de EMLcells do mouse. Estes fatores de transcrição pode desempenhar um papel importante na determinação da chave entre as células Lin-CD34 @ auto-renovação Lin-CD34 + e parcialmente diferenciados.
Abstract
As células estaminais hematopoiéticas (HSCs) são utilizados clinicamente para o tratamento de transplante para reconstruir o sistema hematopoiético de um paciente em muitas doenças, tais como leucemia e linfoma. Elucidação dos mecanismos que controlam HSCs auto-renovação e diferenciação é importante para a aplicação de HSCs para a pesquisa e usos clínicos. No entanto, não é possível obter grande quantidade de HSCs, devido à sua incapacidade para proliferar in vitro. Para superar este obstáculo, foi utilizada uma linha de células derivadas da medula óssea do rato, a linhagem de células EML (eritróide, mielóide e linfóide), como um sistema modelo para este estudo.
RNA-seqüenciamento (RNA-Seq) tem sido cada vez mais usado para substituir microarray para estudos de expressão gênica. Nós relatamos aqui um método detalhado do uso da tecnologia de RNA-Seq para investigar os possíveis fatores-chave na regulação da célula EML auto-renovação e diferenciação. O protocolo fornecido neste artigo está dividido em três partes. O primeiro part explica como a cultura de células de EML e separado Lin-CD34 + e células Lin-CD34 @. A segunda parte do protocolo oferece procedimentos detalhados para a preparação total de RNA ea construção da biblioteca para posterior de alto rendimento de sequenciamento. A última parte descreve o método para análise de dados de RNA-Seq e explica como usar os dados para identificar fatores de transcrição diferencialmente expressos entre Lin-CD34 + e células Lin-CD34 @. Os fatores de transcrição mais significativamente diferencialmente expressos foram identificados como sendo os possíveis reguladores chave que controlam as células EML auto-renovação e diferenciação. Na discussão deste trabalho, destacamos as principais etapas para um desempenho de sucesso desta experiência.
Em resumo, este trabalho oferece um método de usar a tecnologia de RNA-Seq para identificar potenciais reguladores de auto-renovação e diferenciação em células EML. Os principais fatores identificados são submetidos à análise funcional jusante in vitro e in vivo.
Introduction
As células estaminais hematopoiéticas são células sanguíneas raras que residem principalmente no nicho da medula óssea de adultos. Eles são responsáveis pela produção de células necessárias para a reconstituição do sangue e os sistemas imunitários 1. Como um tipo de células estaminais, HSCs são capazes de tanto a auto-renovação e diferenciação. Elucidar os mecanismos que controlam a decisão destino de HSCs, direção ou auto-renovação ou diferenciação, vai oferecer orientações valiosas sobre a manipulação de HSCs para pesquisas de doenças de sangue e uso clínico 2. Um problema enfrentado pelos pesquisadores é que HSCs podem ser mantidas e expandidas in vitro de forma muito limitada; a grande maioria dos seus descendentes são parcialmente diferenciadas em cultura 2.
A fim de identificar os principais reguladores que controlam os processos de auto-renovação e diferenciação em uma escala de todo o genoma, foi utilizada uma linha primitiva de células progenitoras hematopoiéticas do rato EML como um sistema modelo. Thé A linha celular foi derivada a partir de 3,4 medula óssea de murino. Quando alimentados com diferentes factores de crescimento, as células podem diferenciar-se em EML eritróide, mielóide, e células linfóides in vitro, 5. É importante notar que esta linhagem celular pode ser propagada em grande quantidade no meio de cultura contendo factor de células estaminais (SCF) e ainda reter a sua multipotencialidade. Células EML podem ser separados em subpopulações de auto-renovação Lin-SCA + CD34 + e parcialmente diferenciadas células Lin-SCA-CD34 @ com base em marcadores de superfície CD34 e SCA 6. Semelhante a curto prazo HSCs, SCA + CD34 + células são capazes de auto-renovação. Quando tratados com células CD34 + SCF, Lin-SCA + pode regenerar rapidamente uma população mista de células CD34 + e Lin-SCA-CD34 @ Lin-SCA + e continuam a proliferar 6. As duas populações são semelhantes na morfologia e têm níveis semelhantes de ARNm de c-kit e proteína 6. Células Lin-SCA-CD34 @ são capazes de propagar em meios contendo IL-3 em vez de SCF 3. Unveiling principais reguladores da EML decisão destino celular vai oferecer melhor compreensão dos mecanismos celulares e moleculares em transição inicial de desenvolvimento durante a hematopoiese.
A fim de investigar as diferenças moleculares subjacentes entre a auto-renovação de células Lin-SCA-CD34 @ parcialmente diferenciados Lin-SCA + CD34 + e, usamos RNA-Seq para identificar genes diferencialmente expressos. Em particular, vamos nos concentrar em fatores de transcrição, como fatores de transcrição são cruciais na determinação do destino da célula. RNA-Seq é uma abordagem desenvolvida recentemente, que utiliza as capacidades de sequenciamento de próxima geração (NGS) tecnologias para o perfil e quantificar RNAs transcritos do genoma 7,8. Em resumo, o ARN total é poli-A e fragmentada seleccionado como o modelo template.The ARN inicial é então convertido em cDNA usando transcriptase reversa. De modo a mapear full-length transcritos de ARN, utilizando, ARN não degradado intacta para a construção da biblioteca de cDNA é importante. Para o purrepresentar de sequenciamento, as sequências adaptadoras específicos são adicionados a ambas as extremidades do cDNA. Em seguida, na maioria dos casos, as moléculas de ADNc são amplificados por PCR e sequenciado de uma forma de alto rendimento.
Após o seqüenciamento, a resultante leituras podem ser alinhados com um genoma de referência e um banco de dados de transcriptoma. O número de leituras que mapeiam para o gene de referência é contada e essa informação pode ser usada para estimar o nível de expressão do gene. A lê também podem ser montados, de novo sem um genoma de referência, permitindo que o estudo de transcriptomes em organismos modelo não-9. Tecnologia de RNA-seq também tem sido usado para detectar as isoformas de splicing, 10-12 novos transcritos 13 e 14 fusões de genes. Em adição à detecção de genes codificadores de proteínas, o RNA-Seq também pode ser usada para detectar e analisar romance nível de transcrição de RNAs não-codificantes, tais como longa não codificante de ARN 15,16, microARN 17, 18, etc siRNA. Por causa de tele precisão deste método, tem sido utilizado para a detecção de variações de nucleótidos simples 19,20.
Antes do advento da tecnologia de RNA-Seq, microarray foi o principal método utilizado para analisar o perfil de expressão do gene. Pré-concebidas sondas são sintetizados e subsequentemente ligado a uma superfície sólida para formar uma lâmina de microarray 21. ARNm é extraído e convertido em cDNA. Durante o processo de transcrição reversa, os nucleótidos marcados com fluorescência são incorporados no cDNA e o cDNA pode ser hibridado nas lâminas de microarray. A intensidade do sinal recolhido a partir de um ponto específico depende da quantidade de ADNc de ligação com a sonda específica naquele ponto 21. Em comparação com a tecnologia de RNA-Seq, microarray tem várias limitações. Primeiro, microarray confia no conhecimento pré-existente de anotação de genes, enquanto a tecnologia de RNA-Seq é capaz de detectar novas transcrições em alto nível de fundo relativa, o que limita a sua utilização quando a GEnível de expressão ne é baixo. Além disso, a tecnologia de RNA-Seq tem muito maior alcance dinâmico de detecção (8.000 vezes) 7, que, devido ao fundo e saturação dos sinais, a precisão de microarray é limitado para ambos os genes altamente expressos e humilde 7,22. Finalmente, as sondas de microarranjo diferem em suas eficiências de hibridização, que tornam os resultados menos confiáveis quando se comparam os níveis de expressão relativos de diferentes transcritos dentro de uma amostra de 23. Embora RNA-Seq tem muitas vantagens sobre microarranjo, a análise de dados é complexo. Esta é uma das razões que muitos pesquisadores ainda usam microarray em vez de RNA-Seq. Várias ferramentas de bioinformática são necessários para o processamento de dados de RNA-Seq e análise 24.
Entre vários sequenciamento de próxima geração (NGS) plataformas, 454, Illumina, SOLID e Ion Torrent são os mais utilizados. 454 foi a primeira plataforma de NGS comercial. Em contraste com as outras plataformas de sequenciaçãocomprimento como illumina e sólida, a plataforma 454 gera mais ler (em média 700 base de leituras) 25. Lê mais são melhores para a caracterização inicial de transcriptiome devido à sua maior eficiência montar 25. A principal desvantagem da plataforma 454 é o seu custo elevado por megabases de sequência. A Illumina e plataformas SÓLIDOS gerar lê com aumento e comprimentos curtos. O custo por megabases de sequência é muito mais baixa do que a plataforma 454. Devido ao grande número de curta lê para a Illumina e plataformas SÓLIDOS, análise de dados é muito mais computacionalmente intensivo. O preço do instrumento e reagentes para sequenciamento para a plataforma Ion Torrent é mais barato eo tempo de seqüenciamento é mais curto 25. No entanto, a taxa de erro e o custo por megabases de sequência são mais elevados em comparação com o Ilumina e plataformas SÓLIDOS. Plataformas diferentes têm suas próprias vantagens e desvantagens e exigem métodos diferentes para análise de dados. O plaTForm deve ser escolhido com base na finalidade de seqüenciamento e da disponibilidade de financiamento.
Neste artigo, tomamos plataforma Illumina RNA-Seq como um exemplo. Utilizou-se EML célula como um sistema modelo para investigar os reguladores-chave na EML celular auto-renovação e diferenciação, e fornecida uma métodos detalhados de construção da biblioteca de ARN-Seq e análise de dados para o cálculo do nível de expressão e detecção de novo transcrito. Temos mostrado na nossa publicação anterior que estudo RNA-seq no sistema modelo EML 2, quando acoplado com teste funcional (por exemplo, shRNA knockdown) proporcionar uma abordagem poderosa para compreender o mecanismo molecular dos primeiros estágios de diferenciação hematopoiética, e pode servir como um modelo para a análise de células de auto-renovação e diferenciação em geral.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. EML cultura de células e separação das células CD34 + Lin-Lin e-CD34 @ Usar celular Magnetic Classificando Sistema e fluorescência-ativado celular método de classificação
- Preparação de bebê de rim de hamster (BHK) meio de cultura de células-tronco para coleta de fator de célula:
- Cultura de células BHK em meio DMEM contendo 10% de FBS em balão de 25cm 2 (Tabela 1), a 37 ° C, 5% de CO 2 num incubador de cultura de células.
- Quando as células crescem até 80 - 90% de confluência, lave as células uma vez com 10 ml de PBS. Adicionar 5 ml de solução de tripsina-EDTA a 0,25% para a monocamada e incubar as células durante 1-5 minutos à temperatura ambiente (RT) até que as células são separadas.
- Pipeta a solução para cima e para baixo com cuidado para quebrar aglomerados de células. Adicionar 5 ml de DMEM completo ao frasco para parar a actividade de tripsina. Recolher as células por centrifugação a 200 xg durante 5 min à TA.
- Remover o meio e ressuspender o sedimento celular em 10 ml de BHK frescas meio de cultura celular. Transferência de 2 ml da suspensão de células a partir do passo 1.1.4 para um novo frasco de 75 cm2 e adicionar 48 ml de meio de cultura fresca de células BHK para o balão.
- Cultura as células BHK para dois dias e recolher o meio de cultura. A passagem do meio através de um filtro de 0,45 um. Armazenar a forma de -20 ° C até à sua utilização.
- EML cultura de células:
- Culturas de células em suspensão (EML) em meio básico EML contendo meio de cultura de células BHK (Tabela 1), a 37 ° C, 5% de CO 2 num incubador de cultura de células.
- Manter as células EML a baixa densidade celular (0,5-5 x 10 5 células / ml) com o pico de densidade inferior a 6 5 x 10 células / ml. Dividir as células a cada 2-3 dias, na proporção de 1: 5. Células passagem EML suavemente e descartar a cultura após passaging por 10 gerações.
- Depleção de células positivas linhagem:
- Colher as células por centrifugação a EML 200 xg for 5 min e lava-se as células uma vez com PBS. Recolher as células por centrifugação a 200 xg durante 5 min.
- Ressuspender as células com PBS e contagem das células com um hemocitómetro. Determinar a concentração de anticorpos no passo subsequente de separação de células de acordo com o número de células (por favor consulte as instruções proporcionadas pelo fornecedor do sistema de isolamento de células).
- Isolar a linhagem negativa (Lin) usando células de cocktail de anticorpos linhagem (cocktail de anticorpos monoclonais conjugados com biotina-CD5, CD45R (B220), CD11b, Anti-Gr-1 (Ly-6G / C), 7-4 e Ter-119 ) e um sistema de triagem celular activado magnética de acordo com as instruções do fabricante.
- Separação de células CD34 + Lin-e Lin-CD34 @:
- Girar as células Lin @ a partir do passo 1.3.3 a 200 xg durante 5 min. Ressuspender o sedimento de células com PBS e contagem das células com um hemocitómetro.
- Lavar as células duas vezes com tampão FACS e sedimentar as células a 200 xgdurante 5 min.
- Rotular cinco tubos de microcentrífuga de 1,5 ml com o número 1, 2, 3, 4, 5, respectivamente. Ressuspender as células com tampão de SCAF 100 ul por 10 6 células (10 6 células por tubo).
- Adicionar 1 mg de anticorpo anti-CD34 FITC do rato para o tubo 1 e o tubo 2 e misturar os tubos suavemente.
- Incubar todos os tubos a 4 ° C durante 1 hora no escuro.
- Adicionar 0,25 g de anticorpo anti-Sca1 conjugado com PE e 20 ul de anticorpos Linhagem cocktail de APC-conjugado a um tubo, de 0,25 ug de anticorpo conjugado com PE anti-Sca1 de tubo 3, e 20 ul de anticorpos Linhagem cocktail de APC-conjugados aos tubo 4.
- Misturar todos os tubos suavemente e incuba-se as células a 4 ° C durante mais 30 min no escuro.
- Adicionar 300 ul de tampão de FACS e as células para girar para baixo as células a 200 xg durante 5 min.
- Lavam-se as células com 500 ul de tampão de FACS para três vezes.
- Ressuspender o sedimento celular em 500 ul de FACS buffer.
- Use as células em tubos de 2, 3, 4 e 5 para a criação de uma compensação. Isolar células Lin-SCA + CD34 + e Lin-SCA-CD34 @ em tubo 1 usando FACS Aria.
2. RNA Preparação e Biblioteca Construção de alta capacidade Sequencing
- Isolamento, análise de qualidade e quantificação de RNA:
- Extrair o RNA total a partir de Lin-CD34 + e células Lin CD34 @, respectivamente, utilizando-TRIzol seguindo o protocolo fabrica '.
- Remover o ADN utilizando desoxirribonuclease contaminado I (ADNase I), seguindo o protocolo do fabricante. Opcionalmente, o RNA armazenar a -80 ° C nesta etapa para utilização posterior.
- Avaliar a qualidade do RNA total utilizando Bioanalyzer de acordo com as instruções proporcionadas pelo fornecedor. Use amostra de RNA com RNA Integrity Number (RIN) lager do que 9.
- Biblioteca de Construção e de alto rendimento seqüenciamento:
NOTA: Este protocolo descreve RNA-Seq utilizando a plataforma Illumina. Poroutras plataformas de sequenciamento, são necessários diferentes métodos de preparação da biblioteca.- Use 0,1-4 ug de RNA total de alta qualidade por amostra para a preparação da biblioteca. Normalmente 2 ug de RNA total pode ser extraído a partir de células 10 5 EML.
- Use um sistema de preparação de amostras RNA-seqüenciamento para purificação de RNA e fragmentação, primeira e segunda síntese de cDNA, reparação final, 3 'adenilação, adaptador de ligação e amplificação por PCR, seguindo os procedimentos padrões detalhados de instruções do provedor.
- Positivamente seleccionar poliA do mRNA usando oligo-dT esferas magnéticas e fragmentar o ARNm.
- Realizar a transcrição inversa utilizando iniciadores aleatórios para obter o cDNA e subsequentemente sintetizar a segunda cadeia de ADNc para gerar ADNc em cadeia dupla.
- Retirar as saliências 3 'e preencher a 5' salientes por polimerase de DNA. Adenilato extremidades 3 'para evitar a ligação de fragmentos de cDNA a partir de um para o outro.
- Adicionar adaptadores de indexação multiplex para ambas as extremidades do dscDNA. Executar PCR para o enriquecimento de fragmentos de ADN.
- Medir a A260 / A280 para obter informação sobre a concentração da biblioteca utilizando um espectrofotómetro.
- Avaliar a qualidade biblioteca e medir o intervalo de tamanho de fragmentos de ADN utilizando um Bioanalyzer.
Análise 3. Os dados
Para referência do software utilizado nesta parte, consulte (Tabela 2).
- Processamento de arquivos de dados para análise a jusante:
- Converta .bcl (arquivo chamada base) arquivar a .fastq arquivo usando o software casava (Illumina, versão 1.8.2).
- Fogo até o "Terminal" no sistema Linux. Vá para a pasta de dados que contém o arquivo de dados a partir de uma máquina de sequenciamento Illumina HiSeq2000. Suponha que a pasta resultado é 'NASboy1 / JiaqianLabData / HiSeq_RUN / 2013_07_11 / 130627_SN860_0309_A_2013-166_H0PW9ADXX /', tipono comando na Figura S1A, e entrar na pasta de dados.
- Instale casava 1.8.2 no sistema Linux. Suponha que o outputfolder é 'Unaligned ", use o comando na Figura S1B para preparar o arquivo de configuração para a conversão. Use a opção --fastq-cluster-contagem de 0 a garantir apenas um arquivo .fastq é criada para cada amostra. O arquivo gerado é .fastq em formato .gz. Descompactá-lo para análise a jusante (Figura S1B).
- Depois que a pasta 'Unaligned' foi gerado, vá para a pasta 'Unaligned' (Figura S1C).
- Use o comando na Figura S1D para iniciar o processo de conversão. O parâmetro '-j' fornece o número cpu que será usado.
- Depois que o sistema terminar o processo de conversão, vá para a pasta resultado na pasta 'Unaligned' (Figura S1E).
- Use o comando na Figura S1F </ Strong> para descompactar o arquivo .fastq.gz em .fastq arquivo em cada pasta da amostra.
- Converta .bcl (arquivo chamada base) arquivar a .fastq arquivo usando o software casava (Illumina, versão 1.8.2).
- Detectar novas transcrições e avaliar o nível de expressão usando smoking Suíte 26:
- Mapear a-end emparelhado RNA-Seq lê para o genoma de referência do mouse (UCSC versão MM9, obtido a partir http://cufflinks.cbcb.umd.edu/igenomes.html ) usando o software Cartola (versão 1.3.3) 27, que usa o Bowtie ler mapeador (versão 0.12.7) 28. Cartola é fornecido com a opção "-no-novela-juncs" para melhorar a precisão da estimativa do nível de expressão.
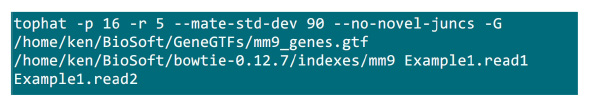
- Coloque os arquivos .fastq em uma pasta onde o processo de mapeamento será implementado. Suponha que há dois arquivos .fastq (renomear a Example1.read1, Example1.read2) para uma amostra de seqüenciamento emparelhado-end, use o comando na Figura S2 para fazer o mapeamento (ajustar os parâmetros de acordo com a configuração do sistema).O parâmetro "-p" fornece o número cpu que será usado. Os parâmetros "-r" e "-mate-std-dev" pode ser obtido a partir da biblioteca QC ou inferidos a partir de um subconjunto de leituras alinhadas (Figura S2).
- Monte o mapeados lê em transcrições de RNA utilizando o software Abotoaduras (versão 1.3.0) 29. Abotoaduras de execução usando o arquivo de anotação de genes conhecidos (mesmo arquivo .gtf usado por Cartola) e arquivo .bam produzido por Cartola.
- Após Cartola terminar a execução, na mesma pasta, use o comando na Figura S3A para executar abotoaduras para construir transcriptoma e estimativa transcrição nível de expressão. O 'mm9_repeatMasker.gtf' e arquivos de seqüência do genoma na pasta 'GenomeSeqMM9' pode ser obtida a partir de UCSC Genome Browser.
- Os arquivos genes.expr e transcripts.expr resultantes contêm o valor de genes e transcrições (isoformas) expressão. Copiar e colaro conteúdo do arquivo para um arquivo do Excel e manipular com aplicativo de planilha (Figura S3B).
- Use o comando na Figura S3C para comparar o arquivo resultante 'transcripts.gtf' para o arquivo de referência 'mm9_genes.gtf', a fim de identificar novos transcrições.
- O arquivo .tmap resultante contém o resultado da comparação. Copie e cole o conteúdo do arquivo em um arquivo de Excel e manipular com aplicativo de planilha. Transcrições com código de classe 'u' pode ser considerado como "romance" em comparação com a referência .gtf arquivo fornecido (Figura S3D).
NOTA: Para a jusante conveniência análise, defina os valores FPKM a 0,1 se os valores estão abaixo de 0,1.
NOTA: Passo 3.2.3 - 3.2.6 é opcional para aqueles que desejam melhorar a precisão da estimativa de expressão novos 'transcrições. Isso vai levar um tempo muito maior, porque o mapeamento e construção transcriptoma precisa ser run mais de uma vez.
- Execute Cartola usando parâmetros padrão e execute abotoaduras para arquivo .gtf gerado usando o comando na Figura S3E.
- Compare o arquivo .gtf resultante para o arquivo .gtf genoma de referência usando o comando na Figura S3F.
- Analisar o arquivo .tmap resultou, conforme descrito na etapa 3.2.2.4. Copie e cole o conteúdo do arquivo em um arquivo de Excel e manipular com aplicativo de planilha. Transcrições com código de classe 'u' pode ser considerado como "romance" em comparação com a referência .gtf arquivo fornecido.
- Após o passo 3.2.5, existe um ficheiro .combined.gtf na pasta que pode ser utilizada como a referência de ficheiro .gtf. Uma segunda execução de Tophat e botão de punho pode ser efectuada tal como descrito no passo 3.2.1 e 3.2.2 para se obter uma estimativa mais precisa da FPKM novos transcritos.
- Mapear a-end emparelhado RNA-Seq lê para o genoma de referência do mouse (UCSC versão MM9, obtido a partir http://cufflinks.cbcb.umd.edu/igenomes.html ) usando o software Cartola (versão 1.3.3) 27, que usa o Bowtie ler mapeador (versão 0.12.7) 28. Cartola é fornecido com a opção "-no-novela-juncs" para melhorar a precisão da estimativa do nível de expressão.
- Detectar differentially expressa genes usando o pacote DESeq 30.
- A entrada de DESeq é uma tabela de contagem de leitura crua. Para obter tal tabela, utilize o script htseq-count distribuído com o pacote HTSeq Python, que pode ser baixado do site HTSeq ( http://www-huber.embl.de/users/anders/HTSeq/doc/count.html ) .
- Certifique-se de que samtools, Python e htseq-count programsare instalado no sistema. Obter números de contagem de leitura brutos de saída cartola usando o comando na Figura S4A.
- Prepare 'Raw_Count_Table.txt', arquivos 'ExperimentDesign.txt' usando o Excel. Copie e salve o conteúdo em formato .txt para o pacote DESeq R (Figura S4B).
- Instalar programa de I no sistema. No terminal, digite "R" e pressione ENTER.A mensagem tela appearas mostrado na Figura S4C.
- Leia 'Raw_Count_Table.txt ',' ExperimentDesign.txt 'em R usando o comando na Figura S4D.
- Carregar pacote DESeq usando o comando na Figura S4E.
- Condições fatorizar em R (Figura S4F).
- Use o comando na Figura S4G para executar o teste binomial negativo na tabela de contagem normalizada.
- Use o comando na Figura S4H a saída diferencial significativo genes expressos em um arquivo .csv.
- A entrada de DESeq é uma tabela de contagem de leitura crua. Para obter tal tabela, utilize o script htseq-count distribuído com o pacote HTSeq Python, que pode ser baixado do site HTSeq ( http://www-huber.embl.de/users/anders/HTSeq/doc/count.html ) .
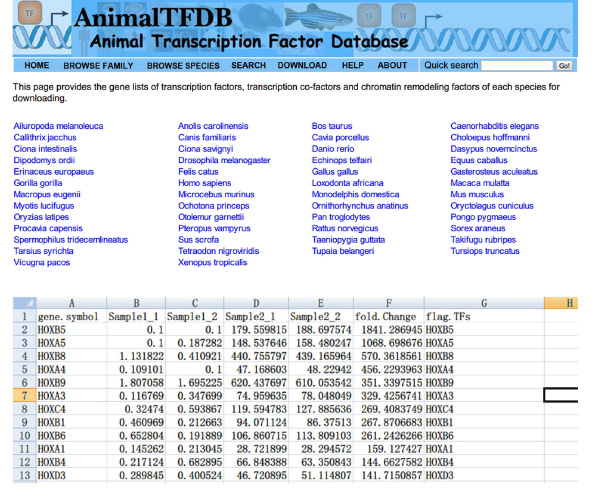
- Fatores de transcrição Lookup "(TFS) valores FPKM toda amostras usando Excel. Intersect DE mesa e mesa gene FT. Os genes pertencem a ambos tabela são diferencialmente expressos factores de transcrição.
- Ir para o site http://www.bioguo.org/AnimalTFDB/download.php e baixar os fatores de transcrição. Em seguida, procurar os fatores de transcrição DE no Excel (< strong> Figura S5).
- Gerando arquivo .bigwig para visualização navegador genoma UCSC.
- Baixar pacote de software '' bedtools do site https://github.com/arq5x/bedtools2 e instalar o software no sistema 31. Faça o download das ferramentas UCSC 'bedGraphToBigWig' do site http://hgdownload.cse.ucsc.edu/admin/exe/ e instalar o software no sistema.
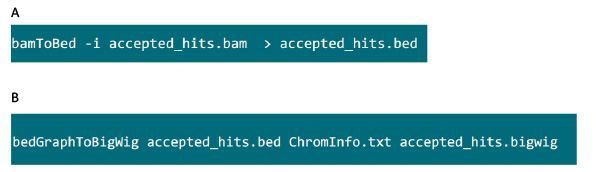
- Na pasta que contém o arquivo .bam, use o comando na Figura S6A para converter arquivos .bam gerado pelo cartola em arquivo .Cama.
- Depois que o arquivo .Cama é produzido, use o comando na Figura S6B para gerar arquivo .bigwig. O arquivo 'ChromInfo.txt' pode ser obtida no seguinte endereço:Arget = "_blank"> http://hgdownload.cse.ucsc.edu/goldenPath/mm9/database/chromInfo.txt.gz.
- Observar uma faixa personalizada no UCSC Genome Browser. Consulte o site http://genome.ucsc.edu/goldenPath/help/customTrack.html sobre como exibir uma faixa personalizada usando o navegador genoma UCSC.

Figura S1: Convertendo arquivo .bcl para .fastq arquivo usando o software casava.

Figura S2: Mapeamento lê a referência genoma usando Cartola.
Figura S3: Detecção de novas transcrições e estimativa do nível de expressão.

Figura S4: Chamando diferencial de genes expressos usando DESeq pacote.

Figura S5: Identificação de fatores de transcrição diferencialmente expressos.

Figura S6: Convertendo resultado do mapeamento para a visualização de dados.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
A fim de analisar os genes diferencialmente expressos em Lin-CD34 + e CD34- células Lin-EML, usamos a tecnologia de RNA-Seq. A Figura 1 mostra o fluxo dos procedimentos. Após o isolamento das células de linhagem negativos por separação de células magnéticas, nós separamos as células Lin-SCA-CD34 @ usando FACS Aria Lin-SCA + CD34 + e. Células EML Lin enriquecidas foram marcadas com anti-CD34, anti-Sca1 e anticorpos cocktail de linhagem. Apenas as células Lin- foram fechado para a análise de Sca1 e CD34 expressão. Duas populações (SCA + CD34 + e CD34- células SCA-EML) poderia ser observado por análise de FACS (Figura 2) 6.
Após a separação de células, foram extraídas a partir de ARN total de células CD34 + e CD34 @, respectivamente, e analisaram a qualidade do RNA. A precisão dos dados de RNA-Seq depende em larga medida da qualidade da biblioteca de ARN-SEQ e a qualidade do RNA total é vital para a preparação de uma biblioteca de alta qualidade. Alta qualidade da amostra de RNA deve ter um valor OD 260/280 entre 1.8 e 2,0. Além de utilizar o espectrofotómetro, a qualidade do RNA foi ainda avaliado com mais precisão por Bioanalyzer. A Figura 3 mostra o resultado de uma amostra de RNA de alta qualidade com o RIN igual a 9,4. Só de alta qualidade amostra de RNA total, com valor RIN superior a 9 foi utilizada para extração de mRNA e procedimentos de construção da biblioteca subseqüentes.
Ribosomal RNA é o tipo mais abundante no ARN de célula. Actualmente duas estratégias principais, a depleção de rRNA ou positivamente selecção de ARNm poliadenilado (ARNm poli-A), são usados para enriquecimento de RNA alvo antes de a construção da biblioteca. Espécies de ARN poliadenilados não são perdidas durante a selecção de poli-A do ARNm. Em contraste, os métodos de esgotamento rRNA como RiboMinus poderia preservar espécies de RNA não poliadenilados. O objetivo do nosso estudo é procurar genes diferencialmente expressos codificação em dois tipos de células, portanto, usamos o método de poli-A seleção mRNA para o enriquecimento de RNAs alvo antes de constru bibliotecacção. Quando a construção da biblioteca foi terminado, o tamanho dos fragmentos de ADN da biblioteca foi verificada antes da sequenciação utilizando Bioanalyzer. A Figura 4 mostra uma biblioteca de boa qualidade com os picos de tamanho de fragmento de cerca de 300 pb.
No passo subsequente, a biblioteca foi submetido a sequenciação de alto rendimento. Em princípio, o comprimento mais ler será útil para o mapeamento de leitura. Isso pode reduzir a probabilidade de que a leitura é mapeado para múltiplos locais, devido à semelhança entre os genes duplicados ou membros da família de genes. Como as sequências de sequenciação par-finais são de ambas as extremidades dos fragmentos, o comprimento de leitura escolhido deve ser inferior a metade do comprimento de fragmentos média. Se o principal objetivo do experimento é medir o nível em vez de construir estrutura transcrição expressão, single-end ler (75 ou 100 pb) pode reduzir o custo sem perder muita informação. Seqüenciamento emparelhado-end é mais útil para a construção de estrutura de transcrição e mais curtoler comprimento pode ser usado para reduzir o custo. Certamente, quando de financiamento suficiente, comprimento mais lido é o preferido.
Para análise da expressão diferencial, existem muitos outros que DESeq algoritmos alternativos. Há também um incluído no pacote abotoaduras chamado cuffdiff 32. DESeq é um dos genes DE algoritmos de análise de contagem mais amplamente utilizados baseiam. DESeq método é baseado em um modelo de estatísticas bem caracterizado - distribuição binomial negativa. Em nossa experiência, DESeq é mais estável comparar com cuffdiff. As primeiras versões do cuffdiff muitas vezes dão significativamente diferentes números de genes DE. Por isso usamos DESeq para análise DE aqui.
Porque os fatores de transcrição são cruciais para a determinação do destino da célula, enfocamos a transcrição significativamente diferencialmente expressos fatores 33. O FT mudou> 1,5 vezes entre Lin-CD34 + e Lin-CD34- foram encontrados e são mostrados no mapa de calor (Figure 5) 2. Notavelmente, o nível relativo de expressão em células Tcf7-Lin CD34 + é mais do que 100 vezes mais elevada do que nas células Lin-CD34 @. Assim Tcf7 foi escolhido para posterior chip Sequenciamento (imunoprecipitação da cromatina e sequenciamento) análise e teste funcional para confirmar a função Tcf7 's na regulação da EML celular auto-renovação e diferenciação 2.

Figura 1:. Fluxo de trabalho dos processos de Lin-CD34 + e CD34 @ Lin-células foram separadas pelo sistema de separação magnética de células e método de classificação de células activadas por fluorescência. O RNA total foi extraído seguido de purificação de ARNm e construção da biblioteca. Após a análise da qualidade da biblioteca, as amostras foram submetidas a alta taxa de transferência de seqüenciamento. Os dados foram analisados e expressos diferencialmente factores de transcrição Foram identificadas.

Figura 2: A separação de células de 6 células Lin ~ EML foram enriquecidas por separação de células magnético-Lin CD34 + e CD34- Lin-. EML. Células Lin- foram marcadas com anti-CD34, anti-Sca1 e anticorpos mistura linhagem. Células Lin- foram fechado para a expressão de CD34 e Sca1. Populações de células CD34-Lin-SCA- EML-Lin CD34 + SCA + e foram classificados.

Figura 3:. Um representante de alta qualidade da amostra de RNA total A qualidade do RNA total foi avaliada por Bioanalyzer. O RNA Integrity Number é 9,4 (FU, Fluorescência unidades).

Figura 4:. Faixa de tamanho de fragmentos de biblioteca emparelhados-End A Distribuição de tamanho de ADN da biblioteca foi analisada utilizando Bioanalyzer. A maioria dos fragmentos estão dentro da gama de tamanhos de 250-500 pb.

Figura 5: diferencialmente expressos. Fatores de transcrição (> 1,5 vezes) entre as células CD34 + Lin-Lin e células-CD34 @ 2 para cada tipo de célula, dois experimentos independentes foram realizados. Genes regulados são indicados como a cor vermelha e para baixo genes regulados são indicados como de cor verde.
Tabela 1: Amortecedores e cultura de células médiuns.
| Software | Uso | Referência | |||
| BOWTIE 1.2.7 | Usado por Cartola para o mapeamento | [28] | |||
| 1.3.3 Cartola | Mapeamento lê volta ao genoma de referência | [27] | |||
| Abotoaduras 1.3.0 | Transcrições de construção e estimativa do nível de expressão | [29] | |||
| DESeq 1.16.0 | Análise da expressão diferencial | [30] | Bedtools 2,18 | Converta arquivos .bam em arquivo .Cama | [31] |
| bedGraphToBigWig | Converta arquivos .Cama para .bigwig arquivo | http://genome.ucsc.edu/ |
Tabela 2: Lista de software para análise de dados.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Transcriptoma de mamíferos é muito complexo 34-38. Tecnologia de RNA-Seq desempenha um papel cada vez mais importante nos estudos de análise de transcriptoma, novela de detecção de transcrições e único nucleotídeo descoberta variação etc. Ele tem muitas vantagens sobre outros métodos de análise de expressão gênica. Como mencionado na introdução, que supera os artefactos de hibridação de microarray e pode ser utilizado para identificar novos transcritos de de novo. Uma limitação do RNA-seqüenciamento é relativa curta duração leitura comparando com seqüenciamento Sanger. No entanto, com a rápida melhoria da tecnologia de sequenciamento, leia comprimento está aumentando constantemente. Neste artigo, nós fornecemos métodos detalhados de usar esta tecnologia para identificar possíveis reguladores chave no rato célula EML auto-renovação e diferenciação.
O primeiro passo fundamental para este protocolo é cultura de células EML. Embora EML é uma linha de células precursoras hematopoiéticas e que podem serpropagadas em grande quantidade com SCF. A condição de cultura de células EML requer mais atenção do que as linhas de células imortalizadas habituais. As células devem ser alimentados e passadas a uma base regular com operação suave; caso contrário, as células podiam alterar nas suas propriedades de auto-renovação e diferenciação e sofrem morte celular. Como o primeiro passo após a recolha de células suficientes, foram isoladas células negativas linhagem usando um sistema de separação de células ativadas magnético. Em seguida, separou as células CD34 @ CD34 + e utilizando separação de células ativadas por fluorescência. As células são normalmente EML passadas menos de 10 gerações antes de utilizar para a extracção de ARN e os números de células CD34 + e células CD34 @ deve ser semelhante após a separação. Se as duas populações variar grandemente em número de células, é aconselhável para descartar a cultura e re-descongelar outro tubo de estoque de células para a cultura.
Após a separação de células CD34 + e células CD34-, extração de RNA total foi realizada, mais um passo importante para este stUdy. RNA de alta qualidade é a base para a construção de uma biblioteca de alta qualidade, que promete a precisão dos dados de sequenciamento. Neste passo crítico, qualquer contacto com RNase devem ser evitados. Todos os reagentes devem ser RNase livre. É importante usar luvas em todos os momentos durante o manuseio de RNA. Alta qualidade amostra de RNA tem um valor OD 260/280 entre 1,8 e 2,0. Ao coletar a fase aquosa contendo o RNA, tenha cuidado para não carregar qualquer fase orgânica com a amostra de RNA. Quaisquer solventes orgânicos residuais, tais como fenol ou clorofórmio no ARN resultaria num valor / 280 DO260 inferior a 1,65. Se o valor DO260 / 280 é menor do que 1,65, o ARN precipitar novamente com etanol. Após lavagem com etanol a 75%, não sedimento de ARN overdry. A secagem sedimento de ARN completamente irá afectar a solubilidade do ARN e levar a um baixo rendimento de ARN.
O próximo passo chave para este protocolo é a preparação da biblioteca. Após extracção de ARN total, utilizando um passo de ADNase para a remoção de ADN contaminado is altamente recomendada, uma vez que a contaminação de ADN pode resultar na estimativa de erro de a quantidade de RNA total usado. Recomenda-se para realizar o procedimento a jusante imediatamente após o isolamento de ARN, uma vez que depois de armazenamento a longo prazo e congelamento-descongelamento procedimento, irá degradar o ARN em algum grau. Se os passos subsequentes após isolamento do ARN não pode ser feita imediatamente, armazenar o ARN em -80 ° C. Antes de RNA total é usado para a purificação de mRNA e síntese de cDNA, a qualidade deve ser sempre verificada. Apenas RNA de alta qualidade pode ser utilizado para a preparação da biblioteca. Usando baixa qualidade ou RNA degradado pode levar a sobre-representação de 3 '. Antes de sequenciamento, foi avaliada a qualidade da biblioteca para garantir a máxima eficiência de seqüenciamento.
Na parte de análise de dados, após a realização de uma corrida de abotoaduras sem um transcriptoma de referência, combinamos as novas transcrições com transcrições conhecidos para formar uma referência .gtf arquivo e executar Cartola e abotoaduras para o segundo tempo.Recomenda-se este procedimento em duas prazo, uma vez que este fornecer estimativa FPKM mais preciso do que correr apenas uma vez. Após análise dos dados, foram identificados os genes diferencialmente expressos. Experimentos a jusante pode ser realizada para validar a função de genes in vitro e in vivo. Em nossa publicação anterior 2, escolhemos os fatores de transcrição significativamente diferencialmente expressos e identificou o local de ligação do genoma desses fatores através da realização de imunoprecipitação da cromatina e seqüenciamento (CHIP-Seq). Além disso, foi aplicado shARN ensaio knockdown para testar o efeito funcional do Tcf7. Nós descobrimos que em células Tcf7 knockdown, os genes foram regulados-se os genes altamente enriquecidas em células CD34 @, enquanto que os genes regulados negativamente foram encontrados para ser significativamente enriquecidos em células CD34 +. Portanto, o perfil de células de knockdown Tcf7 expressão do gene deslocada para uma parcialmente diferenciada CD34- state.Overall, utilizando células EML como um sistema modelojuntamente com a tecnologia de RNA-Seqüenciamento e ensaios funcionais, foram identificados e confirmados Tcf7 como um importante regulador da célula EML auto-renovação e diferenciação.
Subscription Required. Please recommend JoVE to your librarian.
Materials
| Name | Company | Catalog Number | Comments |
| Antibiotic-Antimycotic | Invitrogen | 15240-062 | BHK cell culture |
| Anti-Mouse CD34 FITC | eBioscience | 11-0341-81 | FACS sorting |
| Anti-Mouse Ly-6A/E (Sca-1) PE | eBioscience | 12-5981-81 | FACS sorting |
| APC Mouse Lineage Antibody Cocktail | BD Biosciences | 558074 | FACS sorting |
| BD FACSAria Cell Sorter | BD Biosciences | Special offer sysmtem | FACS sorting |
| Corning™ Cell Culture Treated Flasks 75 cm2 | Corning incorporated | 430641 | Cell culture |
| Corning™ Cell Culture Treated Flasks 25 cm2 | Corning incorporated | 430639 | Cell culture |
| Deoxyribonuclease I, Amplification Grade | Invitrogen | 18068-015 | Library preparation |
| DMEM | Invitrogen | 11965-092 | BHK cell culture |
| DPBS | Gibco | 14190 | Cell culture |
| HI FBS | Invitrogen | 16140071 | BHK cell culture |
| Horse Serum | Invitrogen | 16050-122 | EML cell culture |
| IMDM | HyClone | SH30228.02 | EML cell culture |
| L-Glutamine | Invitrogen | 25030-081 | Cell culture |
| Lineage Cell Depletion Kit, mouse | Miltenyi Biotec | 130-090-858 | Isolation of lineage negative cells |
| NanoVue Plus spectrophotometer | GE Healthcare | 28-9569-62 | Quality control |
| Thermo Scientific™ Napco™ 8000 Water-Jacketed CO2 Incubators | Thermo Scientific | 15-497-002 | Cell culture |
| Penicillin-Streptomycin | Invitrogen | 15140-122 | EML cell culture |
| TRIzol® Reagent | Invitrogen | 15596-018 | RNA exraction |
| TruSeq™ RNA Sample Prep Kit v2 -Set B (48 rxn) | Illumina | RS-122-2002 | Library preparation |
| 2100 Electrophoresis Bioanalyzer Instrument | Agilent | G2939AA | Quality control |
| 0.25% Trypsin-EDTA | Gibco | 25200 | Cell culture |
| 0.45 µm Syringe Filters | Nalgene | 190-2545 | Cell culture |
References
- Chambers, S. M., Goodell, M. A. Hematopoietic stem cell aging: wrinkles in stem cell potential. Stem Cell Rev. 3, 201-211 (2007).
- Wu, J. Q., et al. Tcf7 is an important regulator of the switch of self-renewal and differentiation in a multipotential hematopoietic cell line. PLoS genetics. 8, (2012).
- Ye, Z. J., et al. Complex interactions in EML cell stimulation by stem cell factor and IL-3. Proceedings of the National Academy of Sciences of the United States of America. 108, 4882-4887 (2011).
- Tsai, S., Bartelmez, S., Sitnicka, E., Collins, S. Lymphohematopoietic progenitors immortalized by a retroviral vector harboring a dominant-negative retinoic acid receptor can recapitulate lymphoid, myeloid, and erythroid development. Genes Dev. 8, 2831-2841 (1994).
- Weiler, S. R., et al. D3: a gene induced during myeloid cell differentiation of Linlo c-Kit+ Sca-1(+) progenitor cells. Blood. 93, 527-536 (1999).
- Ye, Z. J., Kluger, Y., Lian, Z., Weissman, S. M. Two types of precursor cells in a multipotential hematopoietic cell line. Proc Natl Acad Sci U S A. 102, 18461-18466 (2005).
- Wang, Z., Gerstein, M., Snyder, M. RNA-Seq: a revolutionary tool for transcriptomics. Nature reviews. Genetics. 10, 57-63 (2009).
- Chu, Y., Corey, D. R. RNA sequencing: platform selection, experimental design, and data interpretation. Nucleic acid therapeutics. 22, 271-274 (2012).
- Hornett, E. A., Wheat, C. W. Quantitative RNA-Seq analysis in non-model species: assessing transcriptome assemblies as a scaffold and the utility of evolutionary divergent genomic reference species. BMC genomics. 13, 361 (2012).
- Eswaran, J., et al. RNA sequencing of cancer reveals novel splicing alterations. Scientific reports. 3, 1689 (2013).
- Wang, E. T., et al. Alternative isoform regulation in human tissue transcriptomes. Nature. 456, 470-476 (2008).
- Wu, J. Q., et al. Dynamic transcriptomes during neural differentiation of human embryonic stem cells revealed by short, long, and paired-end sequencing. Proceedings of the National Academy of Sciences of the United States of America. 107, 5254-5259 (2010).
- Loraine, A. E., McCormick, S., Estrada, A., Patel, K., Qin, P. RNA-seq of Arabidopsis pollen uncovers novel transcription and alternative splicing. Plant physiology. 162, 1092-1109 (2013).
- Edgren, H., et al. Identification of fusion genes in breast cancer by paired-end RNA-sequencing. Genome biology. 12, 6 (2011).
- Ilott, N. E., Ponting, C. P. Predicting long non-coding RNAs using RNA sequencing. Methods. 63, 50-59 (2013).
- Sun, L., et al. Prediction of novel long non-coding RNAs based on RNA-Seq data of mouse Klf1 knockout study. BMC bioinformatics. 13, 331 (2012).
- Luo, S. MicroRNA expression analysis using the Illumina microRNA-Seq Platform. Methods in molecular biology. 822, 183-188 (2012).
- Bolduc, F., Hoareau, C., St-Pierre, P., Perreault, J. P. In-depth sequencing of the siRNAs associated with peach latent mosaic viroid infection. BMC molecular biology. 11, 16 (2010).
- Chepelev, I., Wei, G., Tang, Q., Zhao, K. Detection of single nucleotide variations in expressed exons of the human genome using RNA-Seq. Nucleic acids research. 37, 106 (2009).
- Djari, A., et al. Gene-based single nucleotide polymorphism discovery in bovine muscle using next-generation transcriptomic sequencing. BMC genomics. 14, 307 (2013).
- Murphy, D. Gene expression studies using microarrays: principles, problems, and prospects. Advances in physiology education. 26, 256-270 (2002).
- Chen, K., et al. RNA-seq characterization of spinal cord injury transcriptome in acute/subacute phases: a resource for understanding the pathology at the systems level. PLoS one. 8, 72567 (2013).
- Marioni, J. C., Mason, C. E., Mane, S. M., Stephens, M., Gilad, Y. RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome research. 18, 1509-1517 (2008).
- Ramskold, D., Kavak, E., Sandberg, R. How to analyze gene expression using RNA-sequencing data. Methods in molecular biology. 802, 259-274 (2012).
- Glenn, T. C. Field guide to next-generation DNA sequencers. Mol Ecol Resour. 11, 759-769 (2011).
- Trapnell, C., et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nature protocols. 7, 562-578 (2012).
- Trapnell, C., Pachter, L., Salzberg, S. L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 25, 1105-1111 (2009).
- Langmead, B., Trapnell, C., Pop, M., Salzberg, S. L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome biology. 10, 25 (2009).
- Trapnell, C., et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature. 28, 511-515 (2010).
- Anders, S., Huber, W. Differential expression analysis for sequence count data. Genome biology. 11, 106 (2010).
- Quinlan, A. R., Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 26, 841-842 (2010).
- Cheranova, D., et al. RNA-seq analysis of transcriptomes in thrombin-treated and control human pulmonary microvascular endothelial cells. J Vis Exp. , (2013).
- Zhang, H. M., et al. AnimalTFDB: a comprehensive animal transcription factor database. Nucleic acids research. 40, 144-149 (2012).
- Wu, J. Q., et al. Systematic analysis of transcribed loci in ENCODE regions using RACE sequencing reveals extensive transcription in the human genome. Genome Biol. 9, 3 (2008).
- Wu, J. Q., et al. Large-scale RT-PCR recovery of full-length cDNA clones. Biotechniques. 36, 690-696 (2004).
- Wu, J. Q., Shteynberg, D., Arumugam, M., Gibbs, R. A., Brent, M. R. Identification of rat genes by TWINSCAN gene prediction, RT-PCR, and direct sequencing. Genome Res. 14, 665-671 (2004).
- Dewey, C., et al. Accurate identification of novel human genes through simultaneous gene prediction in human, mouse, and rat. Genome Res. 14, 661-664 (2004).
- Wu, J. Characterize Mammalian Transcriptome Complexity. , LAP Lambert Academic Publishing. (2011).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}