

Summary
RNA-sekvensering och bioinformatik analyser användes för att identifiera väsentligen och differentiellt uttryckta transkriptionsfaktorer i Lin-CD34 + och Lin-CD34- subpopulationer av mus EMLcells. Dessa transkriptionsfaktorer kan spela viktiga roller i att bestämma växla mellan självförnyande Lin-CD34 + och delvis differentierade Lin-CD34- celler.
Abstract
Hematopoetiska stamceller (HSCs) används kliniskt för transplantation behandling för att bygga om en patients hematopoietiska system i många sjukdomar som leukemi och lymfom. Belysa de mekanismer som styr HSCs självförnyelse och differentiering är viktigt för tillämpningen av HSCs för forskning och klinisk användning. Det är emellertid inte möjligt att erhålla stora mängder av HSC: er på grund av deras oförmåga att proliferera in vitro. För att övervinna detta hinder, använde vi en mus-benmärg härledd cellinje, EML (erytroid, myeloid och lymfatisk) cellinjen, som ett modellsystem för denna studie.
RNA-sekvensering (RNA-Seq) har blivit allt vanligare att ersätta microarray för genuttryck studier. Vi redovisar här en detaljerad metod för att med hjälp av RNA-Seq teknik för att undersöka de potentiella nyckelfaktorer reglering av EML cellsjälvförnyelse och differentiering. Protokollet lämnas i detta dokument är uppdelat i tre delar. Den första parit förklarar hur man kultur EML celler och separat Lin-CD34 + och Lin-CD34- celler. Den andra delen av protokollet ger detaljerade förfaranden för total RNA förberedelser och den efterföljande bibliotekskonstruktion för hög genomströmning sekvensering. Den sista delen beskriver metoden för RNA-Seq dataanalys och förklarar hur du kan använda informationen för att identifiera differentiellt uttryckta transkriptionsfaktorer mellan Lin-CD34 + och Lin-CD34- celler. De mest betydande differentiellt uttryckta transkriptionsfaktorer identifierades som potentiella nyckelregulatorer styr EML cellsjälvförnyelse och differentiering. I diskussionsavsnittet i detta dokument lyfter vi de viktigaste stegen för framgångsrikt utförande av detta experiment.
Sammanfattningsvis erbjuder detta papper en metod att använda RNA-Seq teknik för att identifiera potentiella regulatorer av självförnyelse och differentiering i EML-celler. De viktigaste faktorerna som identifierats utsätts för nedströms funktionell analys in vitro och jagn vivo.
Introduction
Hematopoetiska stamceller är sällsynta blodkroppar som är bosatta främst i vuxen benmärg nisch. De är ansvariga för produktionen av celler som krävs för att fylla blodet och immunsystem 1. Som ett slags stamceller, HSCs klarar av både självförnyelse och differentiering. Belysa mekanismer som styr ödet beslut HSCs, mot antingen självförnyelse eller differentiering, kommer att erbjuda värdefull vägledning om manipulation av HSCs för blodsjukdomar undersökningar och klinisk användning 2. Ett problem som forskarna står inför är att HSCs kan vidmakthållas och utvecklas in vitro i mycket begränsad omfattning; de allra flesta av deras avkommor är delvis differentieras i kultur 2.
För att identifiera viktiga regulatorer som styr processerna för självförnyelse och differentiering i genomet hela skalan, använde vi en mus primitiv hematopoetisk stamcellstransplantation linje EML som modellsystem. Thär cellinjen härleddes från murin benmärg 3,4. Då matas med olika tillväxtfaktorer, kan EML celler differentierar till erytroid, myeloid och lymfoida celler in vitro 5. Viktigt kan denna cellinje förökas i stora mängder i odlingsmedium innehållande stamcellsfaktor (SCF) och ändå behålla sin multipotentialitet. EML-celler kan separeras i underpopulationer av självförnyande Lin-SCA + CD34 + och delvis differentierade Lin-SCA-CD34- celler baserat på ytmarkörer CD34 och SCA 6. I likhet med kortsiktiga HSCs, SCA + CD34 + celler kan självförnyelse. Vid behandling med SCF, Lin-SCA + CD34 + celler kan snabbt regenerera en blandad population av Lin-SCA + CD34 + och Lin-SCA-CD34- cellerna och fortsätter att föröka 6. De två populationerna är av samma morfologi och har liknande nivåer av c-kit-mRNA och protein 6. Lin-SCA-CD34- celler är kapabla av föröknings i media innehållande IL-3 i stället för SCF 3. Unveiling nyckelregulatorer i EML cellöde beslut kommer att erbjuda bättre förståelse av cellulära och molekylära mekanismer i början utvecklings övergång under blodbildningen.
För att undersöka de bakomliggande molekylära skillnader mellan självförnyande Lin-SCA + CD34 + och delvis differentierade Lin-SCA-CD34- celler använde vi RNA-Seq för att identifiera differentiellt uttryckta gener. Framför allt fokuserar vi på transkriptionsfaktorer, som transkriptionsfaktorer är avgörande för cell öde. RNA-Seq är en nyligen utvecklad metod som utnyttjar funktionerna i nästa generations sekvensering (NGS) teknik för att profilera och kvantifiera RNA transkriberas från genomet 7,8. I korthet är den totala RNA-poly-A-vald och splittrat som den initiala template.The RNA-mallen omvandlas sedan till cDNA med användning av omvänt transkriptas. För att kart fullängd RNA-transkript, med användning av intakta, icke-nedbruten RNA för konstruktion av cDNA-biblioteket är viktig. För purposerar sekvense de specifika adaptersekvenser läggs till båda ändarna av cDNA. Sedan, i de flesta fall, är cDNA-molekyler amplifieras genom PCR och sekvenserades i en hög genomströmning sätt.
Efter sekvensering, läser den resulterande kan anpassas till en referens genom och en transkriptom databas. Antalet läser den kartan med referensgenen räknas och denna information kan användas för att uppskatta genuttryck nivån. Den läser kan även monteras de novo utan referens genomet, vilket möjliggör studier av transcriptomes i icke-modellorganismer 9. RNA-seq teknik har också använts för att detektera skarv isoformer 10-12, nya avskrifter 13 och genfusioner 14. Utöver detektion av protein-kodande gener, kan RNA-Seq också användas för att upptäcka nya och analysera transkriptionsnivå av icke-kodande RNA, såsom långa icke-kodande RNA 15,16, mikroRNA 17 siRNA etc. 18. På grund av than riktigheten i denna metod har det använts för detektering av enstaka nukleotidvariationer 19,20.
Före tillkomsten av RNA-Seq teknik, microarray var den huvudsakliga metoden som används för att analysera genuttryck profil. Fördesignade prober syntetiseras och därefter bunden till en fast yta för att bilda en microarray bild 21. mRNA extraheras och omvandlas till cDNA. Under den omvända transkriptionsprocessen, är fluorescensmärkta nukleotider införlivas i cDNA: t och cDNA: t kan hybridiseras till de microarray slides. Intensiteten hos signalen som samlats in från en specifik plats är beroende av mängden av cDNA-bindning till den specifika sonden på den platsen 21. Jämfört med RNA-Seq teknik, har microarray flera begränsningar. Först bygger microarray på redan existerande kunskap om gen annotation, medan RNA-Seq teknik kan upptäcka nya transkript vid relativt hög bakgrundsnivå, vilket begränsar dess användning när GEne uttrycksnivån är låg. Dessutom har RNA-Seq tekniken mycket högre dynamiskt omfång detektions (8.000 gånger) 7, medan, beroende på bakgrund och mättnad av signaler, är begränsat både högt och lågt uttryckta gener 7,22 noggrannhet microarray. Slutligen, microarray sonder skiljer sig i sina hybridiseringsförfaranden effektivitet, vilket gör resultaten mindre pålitliga när man jämför de relativa expressionsnivåer av olika transkript i ett prov 23. Även RNA-Seq har många fördelar över microarray, är dess dataanalys komplex. Detta är en av anledningarna till att många forskare fortfarande använder mikromatris istället för RNA-Seq. Olika bioinformatiska verktyg behövs för RNA-Seq databehandling och analys 24.
Bland flera nästa generations sekvensering (NGS) plattformar, 454, Illumina, SOLID och Ion Torrent är de mest använda sådana. 454 var den första kommersiella NGS plattform. I motsats till de övriga sekvense plattformarsåsom Illumina och SOLID, genererar 454-plattformen längre läsa längd (i genomsnitt 700 bas läser) 25. Längre läser är bättre för första karakterisering av transcriptiome grund av deras högre montera verkningsgrad 25. Den huvudsakliga nackdelen med den 454-plattformen är dess höga kostnad per megabas av sekvensen. Den Illumina och SOLID plattformar genererar läser med ökat antal och längd. Kostnaden per megabas av sekvensen är mycket lägre än den 454-plattformen. På grund av det stora antalet kort läser för Illumina och SOLID plattformar, är dataanalys mycket mer beräkningsintensiv. Priset på instrumentet och reagens för sekvensering för Ion Torrent-plattformen är billigare och sekvenstiden är kortare 25. Men felprocenten och kostnaden per megabas av sekvensen är högre jämfört med Illumina och SOLID plattformar. Olika plattformar har sina egna fördelar och nackdelar, och kräver olika metoder för dataanalys. PLAtform bör väljas utifrån sekvense syfte och tillgången på finansiering.
I detta dokument tar vi Illumina RNA-Seq-plattform som ett exempel. Vi använde EML cell som modellsystem för att undersöka de centrala tillsynsmyndigheterna i EML-cell självförnyelse och differentiering, och som en detaljerad metoder för RNA-Seq bibliotek konstruktion och analys av data för beräkning uttrycksnivå och ny avskrift upptäckt. Vi har visat i vår tidigare publikation att RNA-seq studie i EML modellsystem 2, när den kombineras med funktionstest (t.ex. shRNA knockdown) ger en kraftfull metod för att förstå den molekylära mekanismen för de tidiga stadierna av hematopoetisk differentiering, och kan fungera som en modell för analys av cellsjälvförnyelse och differentiering i allmänhet.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. EML Cell Culture och Separation av Lin-CD34 + och Lin-CD34- celler med Magnetisk cellsortering System och fluorescens-aktiverad cellsortering Metod
- Framställning av babyhamsternjurceller (BHK) cellodlingsmedium för stamcellsfaktor samling:
- Kultur BHK-celler i DMEM-medium innehållande 10% FBS i 25 cm 2-kolv (tabell 1) vid 37 ° C, 5% CO2 i en cellodlingsinkubator.
- När celler växer till 80-90% sammanflödet, tvätta cellerna en gång med 10 ml PBS. Tillsätt 5 ml 0,25% trypsin-EDTA-lösning till monoskiktet och inkubera cellerna under 1-5 min vid rumstemperatur (RT) till dess att cellerna lösgörs.
- Pipet lösningen upp och ner försiktigt för att bryta upp klumpar av celler. Tillsätt 5 ml fullständigt DMEM till kolven för att stoppa trypsinaktiviteten. Samla cellerna genom centrifugering vid 200 xg under 5 min vid RT.
- Avlägsna mediet och återsuspendera cellpelleten i 10 ml färsk BHK cellodlingsmediet. Överför 2 ml av cellsuspensionen från steg 1.1.4 till en ny 75 cm 2-kolv och till 48 ml rent BHK cellodlingsmedium till kolven.
- Kultur för BHK-celler i två dagar och samla odlingsmediet. Passage mediet genom ett 0,45 pm filter. Lagra det medium i -20 ° C tills vidare användning.
- EML cellodling:
- Kultur EML-celler (i suspension) i EML basiskt medium innehållande BHK-cell-odlingsmedium (tabell 1) vid 37 ° C, 5% CO2 i en cellodlingsinkubator.
- Bibehålla de EML-celler vid låg celltäthet (0,5 till 5 x 10 5 celler / ml) med toppdensitet mindre än 6 x 10 5 celler / ml. Split cellerna var 2-3 dagar i förhållandet 1: 5. Passage EML celler försiktigt och kasta kulturen efter passage i 10 generationer.
- Utarmning av linje positiva celler:
- Skörda EML cellerna genom centrifugering vid 200 xg for 5 min, och tvätta cellerna en gång med PBS. Samla cellerna genom centrifugering vid 200 xg under 5 minuter.
- Resuspendera cellerna med PBS och räkna cellerna med en hemocytometer. Bestämma antikroppskoncentrationen i den efterföljande cellseparationssteget i enlighet med antalet av de celler (vänligen se instruktionerna som erbjuds av leverantören av den cellisolering systemet).
- Isolera härstamning negativa (Lin-) celler med hjälp härstamning antikropps cocktail (cocktail av biotin-konjugerade monoklonala antikroppar CD5, CD45R (B220), CD11b, Anti-Gr-1 (Ly-6G / C), 7-4 och Ter-119 ) och en magnetisk aktiverad cellsorteringssystem i enlighet med tillverkarens instruktioner.
- Separation av Lin-CD34 + och Lin-CD34- celler:
- Spinn ner Lin-celler från steg 1.3.3 vid 200 xg under 5 minuter. Resuspendera cellpelleten med PBS och räkna cellerna med en hemocytometer.
- Tvätta cellerna två gånger med FACS-buffert och pelletera cellerna vid 200 x gunder 5 min.
- Märk fem 1,5 ml mikrorör med nummer 1, 2, 3, 4, 5 resp. Resuspendera cellerna med 100 l FACS buffert per 10 6 celler (10 6 celler per rör).
- Tillsätt 1 pg av anti-mus CD34 FITC antikropp till röret 1 och rör 2 och blanda rören försiktigt.
- Inkubera alla rör vid 4 ° C under 1 timme i mörker.
- Lägg 0,25 | ig av PE-konjugerad anti-Sca1 antikropp och 20 ul av APC-konjugerad Lineage Cocktail antikroppar mot röret 1, 0,25 | ig av PE-konjugerad anti-Sca1 antikropp till röret 3, och 20 ul av APC-konjugerade Lineage Cocktail antikroppar till röret 4.
- Blanda alla rören försiktigt och inkubera cellerna vid 4 ° C under ytterligare 30 minuter i mörker.
- Addera 300 pl av FACS-buffert till cellerna och centrifugera ner cellerna vid 200 xg under 5 minuter.
- Tvätta cellerna med 500 | il FACS-buffert för tre gånger.
- Resuspendera cellpelleten i 500 mikroliter av FACS BUffer.
- Använd cellerna i rören 2, 3, 4 och 5 för att inrätta kompensation. Isolera Lin-SCA + CD34 + och Lin-SCA-CD34- celler i röret 1 med hjälp av FACS Aria.
2. RNA Förberedelse och biblioteks Konstruktion för hög kapacitet sekvense
- Isolering, kvalitetsanalys och kvantifiering av RNA:
- Utdrag totala RNA från Lin-CD34 + och Lin-CD34- celler respektive använder TRIzol efter tillverkar "protokollet.
- Ta bort den kontaminerade DNA med användning av deoxiribonukleas I (DNas I) genom att följa tillverkarens protokoll. Eventuellt lagra RNA vid -80 ° C vid detta steg för ytterligare användning.
- Bedöma kvaliteten på total-RNA med användning av Bioanalyzer enligt instruktionerna som erbjuds av leverantören. Använd RNA-prov med RNA Integrity Number (RIN) lager än 9.
- Bibliotek Konstruktion och hög genomströmning sekvensering:
OBS: Detta protokoll beskriver RNA-Seq använder Illumina plattformen. Förandra sekvense plattformar, olika biblioteksberedningsmetoder krävs.- Använd 0.1-4 pg av hög kvalitet totalt RNA per prov för biblioteks beredning. Normalt 2 | ig av totalt RNA kan extraheras från 10 5 EML celler.
- Använd en RNA-sekvenseprovberedningssystemet för RNA rening och fragmentering, första och andra cDNA-syntes, end reparation, 3 'ändarna adenylering, adapter ligering och PCR-förstärkning, efter de detaljerade standardförfaranden från leverantörens anvisningar.
- Positivt välja PolyA mRNA med hjälp av oligo-dT magnetiska kulor och fragmentera mRNA.
- Utföra omvänd transkription med användning av slumpmässiga primrar för att erhålla den cDNA och därefter syntetisera den andra strängen av cDNA för att alstra dubbelsträngat cDNA.
- Ta bort de 3 'överhäng och fylla den 5' överhäng av DNA-polymeras. Adenylat 3 'ändarna för att förhindra att cDNA-fragment från att ligera till en annan.
- Lägg multiplex indexerings adaptrar till båda ändarna av dscDNA. Utför PCR för anrikning av DNA-fragment.
- Mät A260 / A280 att få information om koncentrationen av biblioteket med hjälp av en spektrofotometer.
- Bedöma bibliotekets kvalitet och mäta storleksintervallet av DNA-fragment med användning av en Bioanalyzer.
3. Dataanalys
För referens av programvara som används i denna del, se (tabell 2).
- Datafil bearbetning för nedströms analys:
- Konvertera .bcl (bas samtals fil) filen till .fastq fil med hjälp CASAVA programvara (Illumina, version 1.8.2).
- Brand upp "Terminal" i Linux-system. Gå till mappen data som innehåller datafilen från en Illumina HiSeq2000 sekvense maskin. Antag resultatet mappen är "NASboy1 / JiaqianLabData / HiSeq_RUN / 2013_07_11 / 130627_SN860_0309_A_2013-166_H0PW9ADXX / ', typi kommandot i Figur S1A och ange datamappen.
- Installera CASAVA 1.8.2 i Linux-system. Antag att outputfolder är "icke-justerade", använd kommandot i Figur S1B att förbereda konfigurationsfilen för konvertering. Använd alternativet --fastq-cluster-count 0 för att säkerställa en enda .fastq fil skapas för varje prov. Den genererade .fastq filen är i .gz format. Packa upp den för nedströms analys (figur S1B).
- Efter den "icke-justerade" mapp har skapats går du till "icke-justerade" mappen (Figur S1C).
- Använd kommandot i Figur S1D att starta konverteringsprocessen. Den "-j 'parametern förser CPU nummer som kommer att användas.
- När systemet är klar konverteringsprocessen, gå till resultatet mapp under "icke-justerade" mappen (Figur S1E).
- Använd kommandot i Figur S1F </ Strong> för att expandera .fastq.gz filen till .fastq filen under varje provmapp.
- Konvertera .bcl (bas samtals fil) filen till .fastq fil med hjälp CASAVA programvara (Illumina, version 1.8.2).
- Upptäcka nya avskrifter och utvärdera uttrycket nivån med Tuxedo Suite 26:
- Kartlägga den parade-end-RNA-Seq läser till referens musgenomet (UCSC version mm9, erhållen från http://cufflinks.cbcb.umd.edu/igenomes.html ) med Tophat (version 1.3.3) 27, som använder den Bowtie läste mapper (version 0.12.7) 28. Tophat levereras med "-no-roman-juncs" alternativet för att förbättra uppskattningen noggrannhet uttrycksnivå.
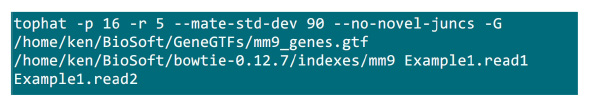
- Placera .fastq filer i en mapp där mappningsprocessen kommer att genomföras. Antag att det finns 2 .fastq filer (byta namn till Example1.read1, Example1.read2) för en parad-end sekvense prov, använd kommandot i figur S2 för att göra kartläggningen (justera parametrarna enligt systeminställningarna).Den "-p" parametern förser CPU nummer som kommer att användas. De "-r" och "-mate-std-dev" parametrar kan erhållas från biblioteks QC eller härledas från en delmängd av linje lyder (figur S2).
- Montera den mappade läser i RNA-transkript med hjälp av Manschettknappar (version 1.3.0) 29. Kör Manschettknappar använder anteckningsfilen kända gener (samma .gtf fil används av Tophat) och .bam fil producerad av Tophat.
- Efter Tophat körts, i samma mapp, använd kommandot i Figur S3A att köra manschettknappar att konstruera transkriptom och uppskattning avskrift uttrycksnivå. Den "mm9_repeatMasker.gtf" och genomsekvensfilerna i "GenomeSeqMM9" mapp kan erhållas från UCSC Genome Browser.
- De resulte genes.expr och transcripts.expr filer innehåller uttrycket värdet av gener och avskrifter (isoformer). Kopiera och klistra infilens innehåll till en Excel-fil och manipulera med kalkylprogram (figur S3B).
- Använd kommandot i Figur S3C att jämföra den resulterande "transcripts.gtf" filen till referensen "mm9_genes.gtf" filen för att identifiera nya avskrifter.
- Den erhållna .tmap filen innehåller jämförelseresultatet. Kopiera och klistra in filens innehåll till en Excel-fil och manipulera med kalkylprogram. Avskrifter med klasskoden 'u' kan betraktas som "nya" jämfört med referens .gtf filen tillgänglig (figur S3D).
OBS: För nedströms analys bekvämlighet, ställ in FPKM värden till 0,1 om värdena är under 0,1.
OBS: Steg 3.2.3 - 3.2.6 är frivilligt för dem som vill förbättra noggrannheten av nya transkript "uttryck uppskattning. Detta kommer att ta mycket längre tid, eftersom kartläggning och transkriptom konstruktion måste vara run mer än en gång.
- Kör Tophat med standardparametrar och sedan köra manschettknappar till genererade .gtf fil med kommandot i Figur S3E.
- Jämför den resulte .gtf filen till referens genomet .gtf fil med kommandot i Figur S3F.
- Tolka result .tmap filen enligt beskrivningen i steg 3.2.2.4. Kopiera och klistra in filens innehåll till en Excel-fil och manipulera med kalkylprogram. Avskrifter med klasskoden 'u' kan betraktas som "nya" jämfört med referens .gtf filen tillhandahålls.
- Efter steg 3.2.5, finns det en .combined.gtf fil i mappen som kan användas som referens .gtf filen. En andra körning av Tophat och manschettknappar kan utföras såsom beskrivits i steg 3.2.1 och 3.2.2 för att erhålla en mer noggrann FPKM uppskattning av nya transkript.
- Kartlägga den parade-end-RNA-Seq läser till referens musgenomet (UCSC version mm9, erhållen från http://cufflinks.cbcb.umd.edu/igenomes.html ) med Tophat (version 1.3.3) 27, som använder den Bowtie läste mapper (version 0.12.7) 28. Tophat levereras med "-no-roman-juncs" alternativet för att förbättra uppskattningen noggrannhet uttrycksnivå.
- Detektera differentially uttryckta gener med användning DESeq paketet 30.
- Inmatningen av DESeq är en rå lästa pulser tabell. För att få en sådan tabell, använd htseq-count skript distribueras med HTSeq Python paket som kan laddas ner från HTSeq webbplats ( http://www-huber.embl.de/users/anders/HTSeq/doc/count.html ) .
- Säkerställa att samtools, Python, och htseq-count programsare installeras i systemet. Skaffa råa siffror lästa räkna från tophat utgång genom att använda kommandot i Figur S4A.
- Förbered 'Raw_Count_Table.txt', 'ExperimentDesign.txt' filer med Excel. Kopiera och spara innehållet i .txt format för DESeq R-paketet (figur s4b).
- Installera R program i systemet. I terminalen, skriv "R" och tryck ENTER.A skärmbild att appearas visade i figur S4C.
- Läs 'Raw_Count_Table.txt ',' ExperimentDesign.txt 'i R med kommandot i Figur S4D.
- Ladda DESeq paketet med kommandot i Figur S4E.
- Faktorisera villkor i R (figur S4F).
- Använd kommandot i Figur S4G att köra negativa binominal test på den normaliserade räkna bordet.
- Använd kommandot i Figur S4H att mata betydande differential uttryckta gener i en CSV-fil.
- Inmatningen av DESeq är en rå lästa pulser tabell. För att få en sådan tabell, använd htseq-count skript distribueras med HTSeq Python paket som kan laddas ner från HTSeq webbplats ( http://www-huber.embl.de/users/anders/HTSeq/doc/count.html ) .
- Lookup transkriptionsfaktorer (TF) FPKM värden i olika prover med Excel. Intersect DE genen bord och TF tabellen. Gener tillhör både tabellen är differentiellt uttryckta transkriptionsfaktorer.
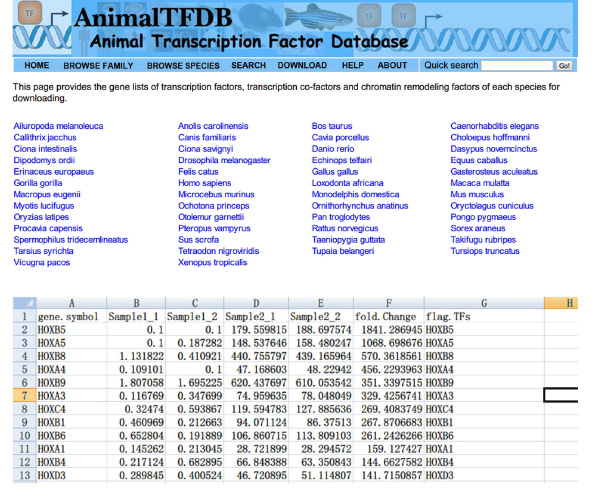
- Gå till webbplatsen http://www.bioguo.org/AnimalTFDB/download.php och ladda ner transkriptionsfaktorer. Slå sedan DE transkriptionsfaktorer i Excel (< strong> Bild S5).
- Generera .bigwig fil för UCSC genomet webbläsare visualisering.
- Ladda ner "bedtools" programpaket från webbplatsen https://github.com/arq5x/bedtools2 och installera programvaran i systemet 31. Ladda ner UCSC Tools bedGraphToBigWig 'från webbplatsen http://hgdownload.cse.ucsc.edu/admin/exe/ och installera programvaran i systemet.
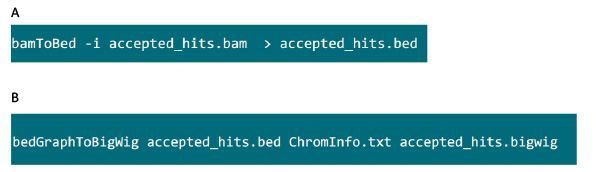
- I mappen som innehåller .bam filen, använder du kommandot i Figur S6A att konvertera .bam fil som genereras av tophat i .bed fil.
- När .bed filen produceras, använd kommandot i Figur S6B generera .bigwig fil. Filen "ChromInfo.txt" kan erhållas från följande adress:Arget = "_blank"> http://hgdownload.cse.ucsc.edu/goldenPath/mm9/database/chromInfo.txt.gz.
- Observera en egen låt på UCSC Genome Browser. Se hemsidan http://genome.ucsc.edu/goldenPath/help/customTrack.html om hur du vill visa en anpassad spår med UCSC genomet webbläsare.

Figur S1: Konvertera .bcl fil till .fastq fil med hjälp CASAVA programvara.

Figur S2: Kartläggning läser till referens genomet med hjälp Tophat.
Bild S3: Upptäckt av nya betyg och uttrycksnivå uppskattning.

Figur S4: Ringa differential uttryckt gen med hjälp DESeq paket.

Figur S5: Identifiering av differentiellt uttryckta transkriptionsfaktorer.

Figur S6: Konvertera kartläggning resultat för datavisualisering.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
För att analysera differentiellt uttryckta gener i Lin-CD34 + och Lin-CD34- EML celler använde vi RNA-Seq teknik. Figur 1 visar arbetsflödet av förfarandena. Efter isolering av härstamnings negativa celler genom magnetisk cellsortering, separerade vi Lin-SCA + CD34 + och Lin-SCA-CD34- celler med hjälp av FACS Aria. Lin-anrikade EML celler färgades med anti-CD34, anti-Sca1 och härstamning cocktail antikroppar. Endast Lin celler gated för analys av Sca1 och CD34 uttryck. Två populationer (SCA + CD34 + och SCA-CD34- EML celler) kan observeras av FACS-analys (figur 2) 6.
Efter cellseparation, vi extraherade total RNA från CD34 + och CD34- celler respektive och analyserat kvaliteten på RNA. Noggrannheten hos RNA-Seq uppgifter bygger till stor del på kvaliteten på RNA-Seq bibliotek och kvaliteten på totalt RNA är avgörande för att förbereda en högkvalitativ bibliotek. Hög kvalitet RNA-prov ska ha ett OD 260/280 värde mellan 1.8 och 2,0. Förutom att med en spektrofotometer, var RNA kvalitet utvärderas ytterligare med större noggrannhet vid Bioanalyzer. Figur 3 visar ett resultat av en hög kvalitet RNA-prov med RIN lika med 9,4. Endast hög kvalitet totalt RNA-prov med RIN värde större än 9 användes för mRNA-extraktion och därpå följande bibliotekskonstruktionsförfaranden.
Ribosomalt RNA är den mest förekommande typen av RNA i cellen. För närvarande två huvudstrategier, utarmning av rRNA eller positivt urval av polyadenylerad mRNA (poly-A mRNA), används för anrikning av mål-RNA före bibliotekskonstruktion. Icke polyadenylerade RNA arter försvinner under valet av poly-A mRNA. I motsats härtill kunde rRNA utarmningsmetoder såsom RiboMinus bevara icke polyadenylerade RNA-species. Syftet med vår studie är att leta efter differentiellt uttryckta kodande gener i två celltyper, vilket vi använde poly-A mRNA urvalsmetod för anrikning av mål-RNA före biblioteks construction. När bibliotekskonstruktion var färdig, var storleken av DNA-fragmenten i biblioteket kontrolleras innan sekvensering med Bioanalyzer, fig. 4 visar en god kvalitet bibliotek med fragment size toppar vid ca 300 bp.
I det efterföljande steget, var biblioteket utsattes för hög genomströmning sekvensering. I princip kommer längre läsa längden vara till hjälp för läsning kartläggning. Det kan minska sannolikheten att läs- mappas till flera platser på grund av likheten mellan dubbla gener eller medlemmar gen familj. Som par-end sekvense sekvenser är från båda ändar av fragmenten bör läslängd valt att vara mindre än hälften av den genomsnittliga fragment längd. Om det huvudsakliga målet med försöket är att mäta uttrycksnivån i stället för att bygga avskrift struktur, single-end läsa (75 eller 100 bp) kan minska kostnaderna utan att förlora för mycket information. Kopplade-end sekvense är mer användbar för avskrift struktur konstruktion och kortareläs längd kan användas för att minska kostnaden. Visst, när tillräckliga medel finns tillgängliga, är längre läses längd föredra.
För differentialexpressionsanalys, det finns många andra än DESeq alternativa algoritmer. Det finns också en inkluderad i manschettknappar paket som heter cuffdiff 32. DESeq är en av de mest använda count baserade DE genanalys algoritmer. DESeq metod är baserad på en väl karakteriserad statistik modell - negativ binomialfördelning. Enligt vår erfarenhet är DESeq stabilare jämfört med cuffdiff. Tidiga versioner av cuffdiff ger ofta väsentligt olika antal DE gener. Därför använde vi DESeq för DE-analys här.
Eftersom transkriptionsfaktorer är avgörande för Determinering har vi fokuserat på den betydligt differentiellt uttryckta transkriptionsfaktorer 33. Den TF bytte> 1,5 gånger mellan Lin-CD34 + och Lin-CD34- hittades och visas på heatmap (Figure 5) 2. Noterbart är den relativa expressionsnivån av Tcf7 i Lin-CD34 + celler mer än 100 gånger högre än den i Lin-CD34- celler. Tcf7 Således valdes för ytterligare Chip-sekvensering (Kromatin Immunoprecipitation och sekvensering) analys och funktionstest för att bekräfta Tcf7: s funktion i regleringen av EML cellsjälvförnyelse och differentiering 2.

Figur 1:. Workflow av de förfaranden som Lin-CD34 + och Lin-CD34- celler separerades genom magnetisk cellseparationssystem och fluorescensaktiverad cellsorteringsmetod. Totalt RNA extraherades, följt av mRNA-rening och konstruktion av bibliotek. Efter analys av bibliotekets kvalitet togs prover utsattes för hög genomströmning sekvensering. Data har analyserats och differentiellt uttryckta transkriptionsfaktorer identifierades.

Figur 2: Separation av Lin-CD34 + och Lin-CD34- EML celler 6 Lin- EML celler anrikade genom magnetisk cellsortering.. Lin celler färgades med anti-CD34, anti-Sca1 och härstamning blandning antikroppar. Lin celler gated för uttryck av CD34 och Sca1. Lin-CD34 + SCA + och Lin-CD34-SCA- EML cellpopulationer sorterades.

Figur 3:. En representant för högkvalitativ total RNA-prov Kvaliteten på totalt RNA bedömdes av Bioanalyzer. RNA Integrity numret är 9,4 (FU, Fluorescence Units).

Figur 4:. Fragment storleksomfånget parade End Biblioteket DNA-storleksfördelningen hos biblioteket analyserades med användning Bioanalyzer. De flesta fragment är inom storleksområdet 250 till 500 bp.

Figur 5:. Differentiellt uttryckta transkriptionsfaktorer (> 1,5 gånger) mellan Lin-CD34 + celler och Lin-CD34- celler 2 För varje celltyp, har två oberoende experiment. Upp-reglerade gener är indikerade som röd färg och ned-reglerade gener är indikerade såsom grön färg.
Tabell 1: Buffertar och Cell odlingsmedier.
| Mjukvara | Användning | Referens | |||
| Bowtie 1.2.7 | Används av Tophat för kartläggning | [28] | |||
| Tophat 1.3.3 | Kartläggning läser tillbaka till referens genomet | [27] | |||
| Manschettknapp 1.3.0 | Avskrifter konstruktion och uttryck nivå uppskattning | [29] | |||
| DESeq 1.16.0 | Differentialexpressionsanalys | [30] | Bedtools 2,18 | Konvertera .bam fil till .bed fil | [31] |
| bedGraphToBigWig | Konvertera .bed fil till .bigwig fil | http://genome.ucsc.edu/ |
Tabell 2: Lista över programvara för dataanalys.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Däggdjur transkriptom är mycket komplex 34-38. RNA-Seq teknik spelar en allt viktigare roll i studierna av transkriptom analys, roman avskrifter upptäckt och enstaka nukleotidvariation upptäckt etc. Det har många fördelar jämfört med andra metoder för genuttryck analys. Som nämndes i inledningen, övervinner det hybridiserings artefakter av microarray och kan användas för att identifiera nya avskrifter de novo. En begränsning av RNA-sekvensering är relativt kort läslängd jämför med Sånger-sekvensering. Men med den snabba förbättringen av sekvenseringsteknologi, läs längd ökar hela tiden. I detta dokument ger vi detaljerade metoder för att använda denna teknik för att identifiera potentiella nyckelregulatorer i mus EML cellsjälvförnyelse och differentiering.
Den första viktigt steg för detta protokoll är EML cellodling. Även EML är en hematopoetisk prekursor cellinje och det kan varafortplantas i stora mängder med SCF. Odlingen skick EML celler kräver mer uppmärksamhet än de vanliga odödliga cellinjer. Cellerna ska matas och passe vid regelbundet med mild drift; annars cellerna skulle kunna ändra i sina egenskaper av självförnyelse och differentiering och genomgår celldöd. Som det första steget efter att samla in tillräckligt med celler, isolerade vi härstamnings negativa celler med användning av en magnetisk aktiverad cellsorteringssystem. Sedan vi separerade CD34 + och CD34- celler med hjälp av fluorescensaktiverad cellsortering. De EML cellerna normalt passe mindre än 10 generationer innan du använder för RNA-extraktion och antalet CD34 + och CD34- celler bör vara liknande efter separation. Om de två populationerna varierar mycket i cellantalet, är det lämpligt att göra sig av kulturen och re-tina ett annat rör av cell lager för kulturen.
Efter separation av CD34 + och CD34- cell, totalt RNA extraktion utförs ytterligare ett viktigt steg för study. Högkvalitets RNA är basen för konstruktion av en hög kvalitet bibliotek, som lovar riktigheten av de sekvenseringsdata. I detta kritiska steg bör all kontakt med RNas undvikas. Alla reagenser ska RNas fritt. Det är viktigt att använda handskar hela tiden vid hantering RNA. Hög kvalitet RNA-prov har ett OD 260/280 värde mellan 1,8 och 2,0. Vid insamlandet av vattenfasen som innehåller RNA, vara noga med att inte bära någon organisk fas med RNA-prov. Eventuella kvarvarande organiska lösningsmedel som fenol eller kloroform i RNA skulle leda till en OD260 / 280 värde som är lägre än 1,65. Om OD260 / 280 värdet är lägre än 1,65, fälla RNA igen med etanol. Efter tvättning med 75% etanol, Låt inte stryk RNA pellet. Torkning RNA pellets helt kommer att påverka lösligheten av RNA och leda till lågt utbyte av RNA.
Nästa viktiga steg för detta protokoll är biblioteks förberedelse. Efter total RNA-extraktion, ett steg för att använda DNas för bortförande av förorenad DNA is rekommenderas, eftersom DNA-kontaminering kan leda till felaktig uppskattning av beloppet av totalt RNA används. Det rekommenderas att utföra nedströms procedur omedelbart efter RNA-isolering, sedan efter långtidslagring och frys-upptiningsförfarande, kommer RNA bryts ned till en viss grad. Om de efterföljande stegen efter RNA isolering inte kan utföras omedelbart, förvara RNA i -80 ° C. Innan total RNA används för mRNA rening och cDNA-syntes, bör alltid kontrolleras kvaliteten. Endast hög kvalitet RNA kan användas för biblioteks beredning. Med hjälp av låg kvalitet eller försämrad RNA kan leda till överrepresentation av 3-ändarna. Före sekvense var biblioteks kvalitet bedöms säkerställa maximal effektivitet sekvensering.
I dataanalysdelen, efter att ha utfört en körning av Manschettknappar utan referens transkriptom, vi kombinerat de nya utskrifter med kända transkript för att bilda en referens .gtf fil och kör Tophat och manschettknappar för andra gången.Detta förfarande i två run rekommenderas, eftersom detta ger mer exakt FPKM uppskattning än att köra endast en gång. Efter dataanalys, var de differentiellt uttryckta gener identifieras. Nedströms experiment kan utföras för att verifiera funktionen av gener in vitro och in vivo. I vår tidigare offentliggörande 2, valde vi de betydligt differentiellt uttryckta transkriptionsfaktorer och identifierat genomet bindningsstället av dessa faktorer genom att utföra kromatin immunoprecipitation och sekvensering (chip-Seq). Dessutom ansökte vi shRNA knockdown-analys för att testa den funktionella effekten av Tcf7. Vi fann att i Tcf7 Knockdown celler, upp-reglerade gener var generna höganrikat i CD34- celler, medan ned-reglerade gener befanns vara avgörande sätt utvecklas i CD34 + celler. Därför genuttryck profil Tcf7 knockdown celler förskjuten mot en partiellt differentierat CD34- state.Overall, med EML cell som ett modellsystemtillsammans med RNA-sekvenseringsteknologi och funktionella analyser, vi identifierade och bekräftade Tcf7 som en viktig regulator av EML cellsjälvförnyelse och differentiering.
Subscription Required. Please recommend JoVE to your librarian.
Materials
| Name | Company | Catalog Number | Comments |
| Antibiotic-Antimycotic | Invitrogen | 15240-062 | BHK cell culture |
| Anti-Mouse CD34 FITC | eBioscience | 11-0341-81 | FACS sorting |
| Anti-Mouse Ly-6A/E (Sca-1) PE | eBioscience | 12-5981-81 | FACS sorting |
| APC Mouse Lineage Antibody Cocktail | BD Biosciences | 558074 | FACS sorting |
| BD FACSAria Cell Sorter | BD Biosciences | Special offer sysmtem | FACS sorting |
| Corning™ Cell Culture Treated Flasks 75 cm2 | Corning incorporated | 430641 | Cell culture |
| Corning™ Cell Culture Treated Flasks 25 cm2 | Corning incorporated | 430639 | Cell culture |
| Deoxyribonuclease I, Amplification Grade | Invitrogen | 18068-015 | Library preparation |
| DMEM | Invitrogen | 11965-092 | BHK cell culture |
| DPBS | Gibco | 14190 | Cell culture |
| HI FBS | Invitrogen | 16140071 | BHK cell culture |
| Horse Serum | Invitrogen | 16050-122 | EML cell culture |
| IMDM | HyClone | SH30228.02 | EML cell culture |
| L-Glutamine | Invitrogen | 25030-081 | Cell culture |
| Lineage Cell Depletion Kit, mouse | Miltenyi Biotec | 130-090-858 | Isolation of lineage negative cells |
| NanoVue Plus spectrophotometer | GE Healthcare | 28-9569-62 | Quality control |
| Thermo Scientific™ Napco™ 8000 Water-Jacketed CO2 Incubators | Thermo Scientific | 15-497-002 | Cell culture |
| Penicillin-Streptomycin | Invitrogen | 15140-122 | EML cell culture |
| TRIzol® Reagent | Invitrogen | 15596-018 | RNA exraction |
| TruSeq™ RNA Sample Prep Kit v2 -Set B (48 rxn) | Illumina | RS-122-2002 | Library preparation |
| 2100 Electrophoresis Bioanalyzer Instrument | Agilent | G2939AA | Quality control |
| 0.25% Trypsin-EDTA | Gibco | 25200 | Cell culture |
| 0.45 µm Syringe Filters | Nalgene | 190-2545 | Cell culture |
References
- Chambers, S. M., Goodell, M. A. Hematopoietic stem cell aging: wrinkles in stem cell potential. Stem Cell Rev. 3, 201-211 (2007).
- Wu, J. Q., et al. Tcf7 is an important regulator of the switch of self-renewal and differentiation in a multipotential hematopoietic cell line. PLoS genetics. 8, (2012).
- Ye, Z. J., et al. Complex interactions in EML cell stimulation by stem cell factor and IL-3. Proceedings of the National Academy of Sciences of the United States of America. 108, 4882-4887 (2011).
- Tsai, S., Bartelmez, S., Sitnicka, E., Collins, S. Lymphohematopoietic progenitors immortalized by a retroviral vector harboring a dominant-negative retinoic acid receptor can recapitulate lymphoid, myeloid, and erythroid development. Genes Dev. 8, 2831-2841 (1994).
- Weiler, S. R., et al. D3: a gene induced during myeloid cell differentiation of Linlo c-Kit+ Sca-1(+) progenitor cells. Blood. 93, 527-536 (1999).
- Ye, Z. J., Kluger, Y., Lian, Z., Weissman, S. M. Two types of precursor cells in a multipotential hematopoietic cell line. Proc Natl Acad Sci U S A. 102, 18461-18466 (2005).
- Wang, Z., Gerstein, M., Snyder, M. RNA-Seq: a revolutionary tool for transcriptomics. Nature reviews. Genetics. 10, 57-63 (2009).
- Chu, Y., Corey, D. R. RNA sequencing: platform selection, experimental design, and data interpretation. Nucleic acid therapeutics. 22, 271-274 (2012).
- Hornett, E. A., Wheat, C. W. Quantitative RNA-Seq analysis in non-model species: assessing transcriptome assemblies as a scaffold and the utility of evolutionary divergent genomic reference species. BMC genomics. 13, 361 (2012).
- Eswaran, J., et al. RNA sequencing of cancer reveals novel splicing alterations. Scientific reports. 3, 1689 (2013).
- Wang, E. T., et al. Alternative isoform regulation in human tissue transcriptomes. Nature. 456, 470-476 (2008).
- Wu, J. Q., et al. Dynamic transcriptomes during neural differentiation of human embryonic stem cells revealed by short, long, and paired-end sequencing. Proceedings of the National Academy of Sciences of the United States of America. 107, 5254-5259 (2010).
- Loraine, A. E., McCormick, S., Estrada, A., Patel, K., Qin, P. RNA-seq of Arabidopsis pollen uncovers novel transcription and alternative splicing. Plant physiology. 162, 1092-1109 (2013).
- Edgren, H., et al. Identification of fusion genes in breast cancer by paired-end RNA-sequencing. Genome biology. 12, 6 (2011).
- Ilott, N. E., Ponting, C. P. Predicting long non-coding RNAs using RNA sequencing. Methods. 63, 50-59 (2013).
- Sun, L., et al. Prediction of novel long non-coding RNAs based on RNA-Seq data of mouse Klf1 knockout study. BMC bioinformatics. 13, 331 (2012).
- Luo, S. MicroRNA expression analysis using the Illumina microRNA-Seq Platform. Methods in molecular biology. 822, 183-188 (2012).
- Bolduc, F., Hoareau, C., St-Pierre, P., Perreault, J. P. In-depth sequencing of the siRNAs associated with peach latent mosaic viroid infection. BMC molecular biology. 11, 16 (2010).
- Chepelev, I., Wei, G., Tang, Q., Zhao, K. Detection of single nucleotide variations in expressed exons of the human genome using RNA-Seq. Nucleic acids research. 37, 106 (2009).
- Djari, A., et al. Gene-based single nucleotide polymorphism discovery in bovine muscle using next-generation transcriptomic sequencing. BMC genomics. 14, 307 (2013).
- Murphy, D. Gene expression studies using microarrays: principles, problems, and prospects. Advances in physiology education. 26, 256-270 (2002).
- Chen, K., et al. RNA-seq characterization of spinal cord injury transcriptome in acute/subacute phases: a resource for understanding the pathology at the systems level. PLoS one. 8, 72567 (2013).
- Marioni, J. C., Mason, C. E., Mane, S. M., Stephens, M., Gilad, Y. RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome research. 18, 1509-1517 (2008).
- Ramskold, D., Kavak, E., Sandberg, R. How to analyze gene expression using RNA-sequencing data. Methods in molecular biology. 802, 259-274 (2012).
- Glenn, T. C. Field guide to next-generation DNA sequencers. Mol Ecol Resour. 11, 759-769 (2011).
- Trapnell, C., et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nature protocols. 7, 562-578 (2012).
- Trapnell, C., Pachter, L., Salzberg, S. L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 25, 1105-1111 (2009).
- Langmead, B., Trapnell, C., Pop, M., Salzberg, S. L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome biology. 10, 25 (2009).
- Trapnell, C., et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature. 28, 511-515 (2010).
- Anders, S., Huber, W. Differential expression analysis for sequence count data. Genome biology. 11, 106 (2010).
- Quinlan, A. R., Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 26, 841-842 (2010).
- Cheranova, D., et al. RNA-seq analysis of transcriptomes in thrombin-treated and control human pulmonary microvascular endothelial cells. J Vis Exp. , (2013).
- Zhang, H. M., et al. AnimalTFDB: a comprehensive animal transcription factor database. Nucleic acids research. 40, 144-149 (2012).
- Wu, J. Q., et al. Systematic analysis of transcribed loci in ENCODE regions using RACE sequencing reveals extensive transcription in the human genome. Genome Biol. 9, 3 (2008).
- Wu, J. Q., et al. Large-scale RT-PCR recovery of full-length cDNA clones. Biotechniques. 36, 690-696 (2004).
- Wu, J. Q., Shteynberg, D., Arumugam, M., Gibbs, R. A., Brent, M. R. Identification of rat genes by TWINSCAN gene prediction, RT-PCR, and direct sequencing. Genome Res. 14, 665-671 (2004).
- Dewey, C., et al. Accurate identification of novel human genes through simultaneous gene prediction in human, mouse, and rat. Genome Res. 14, 661-664 (2004).
- Wu, J. Characterize Mammalian Transcriptome Complexity. , LAP Lambert Academic Publishing. (2011).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}