dGuo was observed to have a retention time of 4.7 min whereas 8-oxo-dGuo had a retention time of approximately 6.4 min (Figure 2A and B). There is about 1,000-fold difference in the peak heights between the two analytes, as seen in Figure 2C. Voltammograms for 8-oxo-dGuo and dGuo were obtained by running standards at a working potential in the range of +0.2 to +1.1 V. The optimum working potential for 8-oxo-dGuo was determined to be +0.5 V, and +0.9 V for dGuo (Figure 3). These potentials are in agreement with other glassy carbon electrodes described in the literature14,15. The limit of detection and dynamic range of 8-oxo-dGuo and dGuo electrochemical detection is reported to be in the femtomole and nanomole range, respectively8,9. Standard curves for dGuo and 8-oxo-dGuo should be run daily to ensure linear detection across a suitable concentration range and proper performance of the instrument (Figure 4). Standard curves are constructed by plotting the peak area as a function of known analyte concentration (Supplementary Figure 2) that allows one to calculate the analyte concentration in the samples from the equation generated from the standard curves.
Complete DNA digestion is required for accurate measurements of 8-oxo-dGua frequency and several genomic DNA digestion methods have been suggested8,15,16. Where necessary, the efficacy of DNA extraction and digestion can be verified using agarose gel electrophoresis (Figure 5A) and HPLC-ED (Figure 5B). Lanes with obvious DNA smear in Figure 5A (e.g., lanes 2, 4, 6, 9, 11, and 13) indicate the presence of undigested DNA while samples with digested DNA fragments run off the gel and were thus undetected. The plateau in Figure 5B indicates that further incubation of DNA with digestion enzymes beyond 1.5 hr does not result in greater amount of dGuo detected, suggesting nearly complete digestion after 1.5 hr. In addition, both agarose gel electrophoresis and HPLC-ED were used to test the utility of the phosphate-based DNA digestion buffer employed here. This buffer is well matched to the HPLC mobile phase and does not give rise to undesirable detector noise attributable to mixing of dissimilar solutions (data not shown). Therefore, the switch from the buffer suggested in the literature (i.e., Tris-HCl8) to sodium phosphate buffer is recommended for this protocol (left versus right side of Figure 5A). Please note that the underlying assumptions for the optimization of DNA digestion were that enzyme inhibition and sample inhomogeneity were negligible and digestion time was the only limiting factor, as stated above. A possibility that incomplete DNA digestion resulted in fragments that are too small to be detected on the agarose gel remains. Even though such a systematic error would affect all samples equally, further experiments (e.g., treatment of cultured cells with a radiolabelled pro-oxidant10) could be conducted to completely rule out such a possibility.
Unlike standard solutions, the digested DNA, whether from isolated cells or mouse tissues, produced several additional peaks (Figure 6A and 6B). These were distant from the 8-oxo-dGuo and dGuo peaks, but spiking experiments as a part of validation exercises outlined in the Discussion are needed to examine if additional peaks (i.e., sample impurity due to matrix effects, for example) interfere with quantification. The 8-oxo-dGuo peak was confirmed by correspondence of the retention time with the standard; and moreover, increase in the 8-oxo-dGuo peak for samples from cells or animals that underwent pro-oxidant treatment (Figure 7). Pro-oxidant KBrO3 participates in one-electron abstraction from guanine that leads to 8-oxo-dGuo formation17. It is noteworthy that in the absence of the pro-oxidant stress, 2.4 molecules of 8-oxo-dGuo per 107 dGuo were detected in A549 cells (Figure 7A). This is comparable to 4.5 molecules of 8-oxo-dGuo/107 dGuo detected in A549 cells observed using an alternative, mass spectrometry-based approach designed to address potential shortcomings of the HPLC-ED method9. DBC treatment (i.e., daily 20.0 mg DBC/kg bw per day for three days) increased 8-oxo-dGuo levels in mouse spleen DNA. This is also an expected result, since ROS is known to be produced during the metabolism of polycyclic aromatic hydrocarbons in vivo8. The relative levels of 8-oxo-dGuo in the spleen of unstressed animals were 8.3-9.4 8-oxo-dGuo/106 dGuo (Figure 7B), which is in excellent agreement with published values of four 8-oxo-dG molecules/106 dGuo measured in murine splenic cells under standard conditions by HPLC-ED18. Figures 6 and 7 show that the levels of 8-oxo-dGuo above the baseline are very small. Thus if DNA is oxidized to lower levels than observed here, the levels of 8-oxo-dGuo may be indistinguishable from baseline, which should be kept in mind by prospective protocol users.

Figure 1. Structure of 8-oxo-dGuo and its tautomer 8-OH-dGuo. Note that 8-oxo-dGuo rather than 8-OH-dGuo is the major tautomer at pH 7.4.
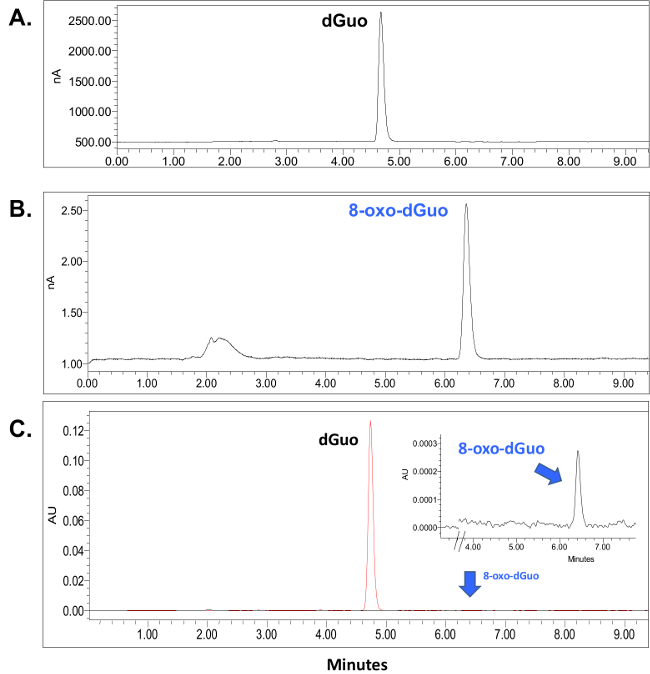
Figure 2. Retention times for dGuo (A) and 8-oxo-dGuo (B). HPLC–ED chromatograms were obtained from a 10.0 μl injection of either dGuo, (1.0 nmol total) or 8-oxo-dGuo (1,000 fmol total) and detected at 0.9 V (A) or 0.5 V (B). The retention peak for dGuo was approximately 4.7 min (A) and that for 8-oxo-dGuo was approximately 6.4 min (B). The broad peak at 2-3 min is likely caused by injection disturbances and is unaffected (i.e., always present at similar intensity) by the concentration of 8-oxo dGuo. (C) Superimposed chromatograms from two separate injections described in (A) and (B) detected by UV (260 nm). The standards were run on the same day using identical run conditions. Please click here to view a larger version of this figure.

Figure 3. Optimizing detector voltage. (A) Voltammogram of dGuo (final concentration 1 nmol) detected by HPLC-ED analysis at a range of 0.7-1.1 V. (B) Voltammogram of 8-oxo-dGuo (final concentration 500 fmol) detected by HPLC-ED analysis at a range of 0.3-0.7 V. Means of three independent experiments (N=3) ± standard deviation shown. In some cases error bars are smaller than the plotting symbols.

Figure 4. Standard curve for dGuo (A) and 8-oxo-dGuo (B). Ten microliters of either dGuo or 8-oxo-dGuo was injected and measured by HPLC-ED at 0.9 V (A) or 0.6 V (B). Means of three independent experiments (N=3) ± standard deviation shown. In some cases error bars are smaller than the plotting symbols.

Figure 5. Optimization of DNA digestion. (A) Eighty microgram of salmon testes DNA was digested as described8 in Tris-HCl, glycine-acetate buffer (left) or sodium phosphate buffer (right). Agarose gel electrophoresis (1.0%) was run at 150 V for 45 min. Lane 1 and 8: DNA ladder; lanes 2, 4, and 6: undigested DNA; lanes 3, 5, and 7: digested DNA; lanes 9, 11, and 13: undigested DNA (phosphate buffer); lanes 10, 12, and 14: digested DNA (phosphate buffer). (B) Salmon testes DNA (80 µg) was digested herein by incubating with DNase I, PDE I, and AP for 0.5-2.5 hr per each step, in 50 mM phosphate buffer containing magnesium chloride and DFO. Fifty microliters of each sample, containing 7.3 µg of digested DNA, was analyzed by HPLC (i.e., 0.9 V and 1 ml/min flow) to detect dGuo. Figure shows means of three independent experiments (N=3) ± standard error of the mean. Please click here to view a larger version of this figure.


Figure 6. Chromatograms for 8-oxo-dGuo detection in cultured cells (A) or animal tissue (B). (A) Digested DNA from A549 cells, treated or untreated with KBrO3. (B) Digested DNA from the spleen of DBC-treated mice. Peaks shifted to the left due to the removal of guard column (due to pressure build-up in the apparatus; their position was confirmed with standards). Standards and samples were run on the same day using identical run conditions and 10 ml injection volumes, at 1 ml/min flow rate and 0.5 and 0.9 V for dGuo and 8-oxo-dGuo, respectively. Please click here for a larger version of panel A; here for panel B.

Figure 7. Quantification of 8-oxo-dGuo in biological samples. Peaks were integrated and compared to standard curves (see Supplementary Figure 2 for more details) to yield 8-oxo-dGuo levels in DNA from A549 cells, treated with KBrO3 (A), or 8-oxo-dGuo levels in DNA from the spleen of DBC-treated mice (B). Stars (*) indicate statistical significance (p < 0.05, one-way analysis of variance (ANOVA) (A), Student’s t-test (B)). Means of three (A) or five (B) independent experiments ± standard error of the mean are shown. Samples were collected 4 or 24 hr after the 3-day treatment.
| Mobile phase | 50.0 mM Na phosphate buffer, pH 6.2, containing 6% MeOH and 2.0 mM KCl |
| Mobile phase flow rate | 1.0 ml/min |
| Column type | YMC-BASIC with bonded spherical silica |
| Column length | 15 cm |
| Column inner diameter | 3.5 mm |
| Column temperature | 35 °C |
| Detector temperature | 29 °C |
| Voltage setting for 8-oxo-dGuo | +0.5 V |
| Voltage setting for dGuo | +0.9 V |
| Injection volume | 10.0 ml |
Table 1. Instrument parameters for detection and quantification of 8-oxo-dGuo by HPLC-ED.
| nmoles | 100 nM standard, µl | HPLC Mobile phase, µl | Dilution, fold |
| 5 | 300 | 0 | 1 |
| 2.5 | 150 | 150 | 2 |
| 1 | 60 | 240 | 5 |
| 0.5 | 30 | 270 | 10 |
| 0.25 | 15 | 285 | 20 |
| 0.1 | 6 | 294 | 50 |
Table 2. Preparation of standard curve for dGuo.
| fmoles | 250 nM standard, µl | HPLC Mobile phase, µl | Dilution, fold |
| 2,500 | 300 | 0 | 1 |
| 1,000 | 120 | 180 | 2.5 |
| 500 | 150 | 150 | 5 |
| 250 | 150 | 150 | 10 |
| 100 | 120 | 180 | 25 |
| 50 | 150 | 150 | 50 |
Table 3. Preparation of standard curve for 8-oxo-dGuo.

Supplementary Figure 1. Comparison of electrochemical (EC) and UV-based detection of dGuo. Standard curve was created for dGuo detection by electrochemical detection (EC) or UV detection (260 nm). Peak areas were integrated for dGuo peak at approximately 4.7 min by both methods. N=3, means ± standard deviation are shown.

Supplementary Figure 2. Quantification of dGuo from its standard curve. An example of the use of standard curve to determine the concentration of dGuo is shown. (A) Peak area can be determined by integration using standard software. (B) This information is used to construct a standard curve in order to use the equation of the line to calculate the unknown analyte concentration in the sample. Please click here to view a larger version of this figure.
