

Summary
Der Vergleich und die Optimierung zweier pflanzenorganischer DNA-Anreicherungsmethoden werden vorgestellt: traditionelle Differentialzentrifugation und Fraktionierung der gesamten gDNA auf Basis des Methylierungsstatus. Wir beurteilen die resultierende DNA-Quantität und -Qualität, zeigen die Leistung in der Kurzzeit-Sequenzierung der nächsten Generation und diskutieren das Potenzial für den Einsatz in der Langzeit-Einzelmolekül-Sequenzierung.
Abstract
Pflanzenorganellare Genome enthalten große, sich wiederholende Elemente, die einer Paarung oder Rekombination unterzogen werden können, um komplexe Strukturen und / oder subgenomische Fragmente zu bilden. Organellare Genome gibt es auch in Beimischungen innerhalb einer gegebenen Zelle oder eines Gewebetyps (Heteroplasmie), und eine Fülle von Subtypen kann sich während der Entwicklung oder unter Belastung (unterstöchiometrische Verschiebung) ändern. Next-Generation-Sequenzierung (NGS) -Technologien sind erforderlich, um ein tieferes Verständnis der organellaren Genomstruktur und -funktion zu erhalten. Traditionelle Sequenzierungsstudien verwenden mehrere Methoden, um organellare DNA zu erhalten: (1) Wenn eine große Menge an Ausgangsgewebe verwendet wird, wird sie homogenisiert und einer differentiellen Zentrifugation und / oder Gradientenreinigung unterworfen. (2) Wenn eine kleinere Menge an Gewebe verwendet wird ( dh wenn Samen, Material oder Raum begrenzt sind), wird das gleiche Verfahren wie in (1) durchgeführt, gefolgt von einer Vollgenom-Amplifikation, um ausreichende DNA zu erhalten. (3) Die Analyse der Bioinformatik kann für die SeqDie gesamte genomische DNA und die Analyse der organellaren liest. Alle diese Methoden haben inhärente Herausforderungen und Kompromisse. In (1) kann es schwierig sein, eine so große Menge an Ausgangsgewebe zu erhalten; In (2) könnte eine Vollgenom-Amplifikation eine Sequenzierungsvorspannung einführen; Und in (3) könnte die Homologie zwischen nuklearen und organellaren Genomen die Versammlung und Analyse beeinträchtigen. In Pflanzen mit großen nuklearen Genomen ist es vorteilhaft, für organellare DNA zu reichern, um Sequenzierungskosten und Sequenzkomplexität für Bioinformatik-Analysen zu reduzieren. Hier vergleichen wir eine traditionelle Differentialzentrifugationsmethode mit einer vierten Methode, einem angepassten CpG-Methyl-Pulldown-Ansatz, um die gesamte genomische DNA in nukleare und organelläre Fraktionen zu trennen. Beide Methoden liefern genügend DNA für NGS, DNA, die für organelläre Sequenzen hoch angereichert ist, wenn auch mit unterschiedlichen Verhältnissen in Mitochondrien und Chloroplasten. Wir stellen die Optimierung dieser Methoden für Weizenblattgewebe vor und diskutieren große Vorteile und dNachfolgen von jedem Ansatz im Rahmen von Stichprobeneingabe, Protokoll-Leichtigkeit und Downstream-Anwendung.
Introduction
Genomsequenzierung ist ein mächtiges Werkzeug, um die zugrunde liegende genetische Basis wichtiger Pflanzenmerkmale zu sezieren. Die meisten Genom-Sequenzierungsstudien konzentrieren sich auf den Kerngenom-Gehalt, da sich die Mehrheit der Gene im Kern befindet. Allerdings organellären Genome, einschließlich der Mitochondrien (über Eukaryonten) und Plastiden (in Pflanzen, die spezielle Form, die Chloroplasten, arbeitet bei der Photosynthese) beitragen signifikante genetische Informationen wesentlich zu organismal Entwicklung, Stressreaktion, und die allgemeine Fitness 1. Organellären Genome werden typischerweise in Gesamt - DNA - Extraktionen , die für nukleare Genom - Sequenzierung enthalten, obwohl Verfahren Organell Zahlen vor der DNA - Extraktion zu reduzieren auch 2 eingesetzt werden. Viele Studien haben Sequenzierungsergebnisse aus Gesamt-gDNA-Extraktionen verwendet, um die organellaren Genome 3 , 4 , 5 ,Xref "> 6 , 7. Wenn jedoch das Ziel der Studie auf organellare Genome zu konzentrieren ist, erhöht die Gesamt-gDNA die Sequenzierungskosten, da viele Lesungen an die nuklearen DNA-Sequenzen, insbesondere in Pflanzen mit großen nuklearen Genomen, verloren gehen Darüber hinaus ist aufgrund der Vervielfältigung und Übertragung von organellaren Sequenzen in das nukleare Genom und zwischen Organellen die Auflösung der korrekten Kartierungsposition der Sequenzierung auf das richtige Genom bioinformatisch anspruchsvoll 2 , 8. Die Reinigung von organellaren Genomen aus dem nuklearen Genom ist eine Strategie, um diese Probleme zu reduzieren. Weitere Bioinformatik-Strategien können verwendet werden, um Lesungen zu trennen, die in Regionen der Homologie zwischen den Mitochondrien und Chloroplasten abbilden.
Während die organellaren Genome aus vielen Pflanzenarten sequenziert wurden, ist wenig über die Breite der organellaren Genomvielfalt bekanntVerfügbar in wilden Populationen oder in kultivierten Zuchtpools. Organellare Genome sind auch als dynamische Moleküle bekannt, die eine signifikante strukturelle Umlagerung durch Rekombination zwischen den Wiederholungssequenzen 9 erfahren. Darüber hinaus sind mehrere Kopien des organellaren Genoms in jeder Organelle enthalten, und mehrere Organellen sind in jeder Zelle enthalten. Nicht alle Kopien dieser Genome sind identisch, was als Heteroplasmie bekannt ist. Im Gegensatz zum kanonischen Bild der "Meisterkreise" gibt es jetzt wachsende Hinweise auf ein komplexeres Bild von organellaren Genomstrukturen, darunter sub-genomische Kreise, lineare Chromosomen, lineare Concatamere und verzweigte Strukturen 10 . Die Zusammenstellung von pflanzlichen organellaren Genomen wird durch ihre relativ großen Größen und erheblichen umgekehrten und direkten Wiederholungen weiter kompliziert.
Traditionelle Protokolle für organellare Isolierung, DNA-Reinigung und nachfolgendes Genom E Sequenzierung sind oft schwerfällig und erfordern große Mengen an Gewebeeintritt, wobei mehrere Gramm bis zu Hunderten von Gramm jungem Blattgewebe als Ausgangspunkt 11 , 12 , 13 , 14 , 15 , 16 , 17 erforderlich sind. Dies macht die organische Genomsequenzierung unzugänglich, wenn das Gewebe begrenzt ist. In einigen Situationen sind die Saatgutmengen begrenzt, z. B. wenn es notwendig ist, auf einer Generationsbasis oder in männlichen sterilen Linien zu sequenzieren, die über eine Kreuzung aufrechterhalten werden müssen. In diesen Situationen kann organellare DNA gereinigt und dann einer Vollgenom-Amplifikation unterworfen werden. Allerdings kann die Vollgenom-Amplifikation eine signifikante Sequenzierungsvorspannung einführen, was ein besonderes Problem bei der Beurteilung von Strukturvariation, sub-genomischen Strukturen und Heteroplasmie-Niveaus ist> 18 Jüngste Fortschritte in der Bibliotheksvorbereitung für Kurz-Lese-Sequenzierungstechnologien haben niedrige Eingangsbarrieren überwunden, um eine Vollgenom-Verstärkung zu vermeiden. Zum Beispiel ermöglicht das Illumina Nextera XT-Bibliotheks-Präparationskit so wenig wie 1 ng DNA als Eingang 19 verwendet werden . Allerdings benötigen Standard-Bibliothekspräparate für Langzeit-Sequenzierungsanwendungen, wie z. B. PacBio- oder Oxford-Nanopore-Sequenztechnologien, noch eine relativ hohe Menge an Input-DNA, die eine Herausforderung für die organelläre Genomsequenzierung darstellen kann. In letzter Zeit wurden neue, benutzerdefinierte, lang ersehnte Sequenzprotokolle entwickelt, um die Eingangsmengen zu reduzieren und zu helfen, die Genomsequenzierung in Proben zu erleichtern, wo das Erhalten von Mikrogrammmengen von DNA schwierig ist 20 , 21 . Allerdings bleibt die Erzielung hochmolekularer, reiner organellarer Fraktionen zur Einspeisung in diese Bibliothekspräparate eine Herausforderung.
Wir suchten tO vergleichen und optimieren organellare DNA Anreicherung und Isolation Methoden geeignet für NGS ohne die Notwendigkeit der Voll-Genom-Amplifikation. Insbesondere war es unser Ziel, Best Practices zu bestimmen, um für hochmolekulare organellare DNA aus begrenzten Ausgangsmaterialien, wie z. B. einer Teilprobe eines Blattes, zu bereichern. Diese Arbeit stellt eine vergleichende Analyse von Methoden zur Anreicherung der organellaren DNA dar: (1) ein modifiziertes, traditionelles Differentialzentrifugationsprotokoll gegenüber (2) einem DNA-Fraktionierungsprotokoll, das auf der Verwendung eines kommerziell erhältlichen DNA-CpG-Methyl-Bindungsdomänen-Protein-Pulldown-Ansatzes basiert 22 auf Pflanzengewebe 23 angewendet. Wir empfehlen Best Practices für die Isolierung von organellaren DNA aus Weizenblattgewebe, die sich leicht auf andere Pflanzen und Gewebetypen erweitern lassen.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. Erzeugung von Pflanzenmaterialien für die organellare Isolierung und DNA-Extraktion
- Standardwachstum von Weizensämlingen
- Pflanzensamen in Vermiculit in kleinen, quadratischen Töpfen mit 4 - 6 Samen pro Ecke. Transfer in eine Gewächshaus- oder Wachstumskammer mit 16 h Lichtkreislauf, 23 ºC Tag / 18 ºC Nacht.
- Wasser die Pflanzen jeden Tag. Befruchten Sie die Pflanzen mit ¼ Teelöffel Granulat 20-20-20 NPK Dünger bei der Keimung und bei 7 Tagen nach der Keimung.
- Alternative Etiolierung von Weizensämlingen
- Folgen Sie Schritt 1.1, aber legen Sie die Töpfe in eine dunkle Wachstumskammer, 23 ° C für 16 h / 18 ºC für 8 h. Alternativ decken Sie die Pflanzen im Gewächshaus ab ( z. B. mit einem Vorratsbehälter, jedoch muss eine ordnungsgemäße Belüftung beibehalten werden).
- Wachstum und Gewebesammlung
- Die Pflanzen für 12 - 14 Tage wachsen lassen. Für die meisten Weizen GenotypS, 75 - 100 Sämlinge ergeben etwa 10 - 12 g Gewebe, das für zwei organellare Extraktionen mit dem Differentialzentrifugationsverfahren (Abschnitt 2) ausreicht; Nur eine Pflanze ist notwendig, wenn man den DNA-CpG-Methylierungs-basierten Pulldown-Ansatz zur Fraktionierung von Organellaren aus nuklearer DNA verwendet (Abschnitt 3).
- Bei Verwendung des Differentialzentrifugationsansatzes sammeln Sie das Gewebe frisch und verfahren Sie sofort mit der Verarbeitung der Proben, wie in Abschnitt 2 beschrieben.
- Bei Verwendung des CpG-Methyl-Pulldown-Ansatzes werden 20 mg Abschnitte des jungen Blattgewebes in Mikrozentrifugenröhrchen geerntet (verwenden Sie entweder Standard-gewachsene oder etiolierte Gewebe, siehe die repräsentativen Ergebnisse ). Snap-Freeze auf flüssigem Stickstoff einfrieren und bei -80 ºC bis zum Gebrauch einfrieren. Fahren Sie mit der Pulldown-Fraktionierung von DNA fort, wie in Abschnitt 3 beschrieben.
2. Methode Nr. 1: DNA-Extraktion mit Differentialzentrifugation (DC)
HINWEIS: Die diffErentliches Zentrifugationsprotokoll wurde aus zwei Publikationen modifiziert, die die Bedingungen optimierten, um beide Organellen zu isolieren, aber für die Mitochondrien 17 , 24 zu bereichern. Das resultierende Protokoll ist weniger zeitintensiv und verwendet weniger toxische Chemikalien als die bisherigen Methoden. Speziell wurden Modifikationen an den Puffern und Waschschritten vorgenommen, einschließlich der Zugabe von Polyvinylpyrrolidon (PVP) zum STE-Extraktionspuffer und die Eliminierung des abschließenden Waschschrittes in NETF-Puffer, der Natriumfluorid (NaF) enthält.
Achtung: Die Vorbereitung und Verwendung von STE-Puffer sollte unter einer chemischen Dunstabzugshaube mit persönlicher Schutzausrüstung durchgeführt werden, da dieser Puffer 2-Mercaptoethanol (BME) enthält.
- Dinge vor dem Start
- Sicherstellen, dass alle Geräte extrem sauber sind und autoklavieren alle Geräte, die autoklaviert werden können ( z. B. Schleifzylinder, High-Speed-CentriFuge Rohre, etc. ).
HINWEIS: Filtertips werden für alle Schritte empfohlen, die ein Pipettieren erfordern, um eine Kreuzkontamination zu vermeiden. - Siehe Liste der benötigten Geräte und Reagenzien und bereiten die erforderlichen Puffer und Arbeitsbestände für Methode Nr. 1 ( Tabelle 1 ) vor. Die kryogenen Schleifblöcke auf -20 ºC und die Rotoren und Puffer auf 4 ºC kühlen, die Mikrozentrifuge auf 4 ºC stellen und ein 37 ºC Wasserbad einschalten.
- Sicherstellen, dass alle Geräte extrem sauber sind und autoklavieren alle Geräte, die autoklaviert werden können ( z. B. Schleifzylinder, High-Speed-CentriFuge Rohre, etc. ).
- Isolierung von Organellen
- 5 g frisches Gewebe ernten und in kaltem, sterilem Wasser in einem gekühlten Becher auf Eis spülen.
HINWEIS: Halten Sie die Proben immer auf Eis während aller Betriebsabläufe und Transporte zu und von den Zentrifugen, Dunstabzugshauben usw. Alternativ arbeiten Sie in einem kalten Raum, wenn der Zugang zu ausreichend Platz und Ausrüstung zur Durchführung des Protokolls besteht. - Mit Scheren schneiden Blatt Blatt in ~ 1-cm-Stücke direkt in ein 50-ml-Rohr mit zwei Keramik-SchleifenZylinder
HINWEIS: Die Schere zwischen den Proben reinigen oder wechseln, um eine Kreuzkontamination zu vermeiden. - Wenn es keinen Gewebehomogenisator gibt, verwenden Sie einen Mörser und eine Pistille und folgen Sie, um die Schritte 2.2.4 - 2.2.9 zu ersetzen.
- Schneide das Blattgewebe in einen vorgekühlten Mörtel auf Eis. Schleifen Sie die Proben für 2 - 3 min in 15 ml STE (in der Dunstabzugshaube).
- Gießen Sie den Puffer (verlassen Sie das Gewebe im Mörser) durch einen Trichter, der eine Schicht von vornassem, sterilem Filtrationstuch (~ 22- bis 25-μm Porengröße, siehe Hauptprotokoll für Details) in ein anderes 50-ml-Rohr enthält . Füge 10 ML STE dem Mörser hinzu und stößt weiter und homogenisiere dich.
- Gießen Sie das homogenisierte Gewebe und Puffer in den gleichen Trichter. Spülen Sie den Mörser und stampfen Sie mit 10 ml STE und gießen Sie ihn in den Trichter. Squeeze und wringe das Filtrationstuch in den Trichter, um so viel Flüssigkeit wie möglich zu erholen.
HINWEIS: Handschuhe zwischen den Proben wechseln, um eine Kreuzkontamination zu vermeiden. Weiter mit dem ProfiTocol in Schritt 2.2.10.
- Füge 20 ml STE (in der Dunstabzugshaube) zu jeder 50 ml Röhre hinzu.
- Legen Sie die Proben in vorgekühlte kryogene Schleifblöcke in eine Gewebe-Schleifvorrichtung und schleifen Sie die Proben für 2 x 30 s bei 1.750 U / min. Drehen Sie die Probenpositionen und stellen Sie die Proben auf Eis für ~ 1 min zwischen den Schleifern.
HINWEIS: In diesem Schritt kann ein Mörtel- und Stößel-, Mischer- oder andere Gewebe-Schleif- / Homogenisierungsgerät verwendet werden. Jedoch wird jede Methode die resultierende DNA-Qualität auf unterschiedliche Grade beeinflussen, und daher sollten DNA-Länge und Qualität beurteilt werden, bevor sie mit nachgeschalteten Anwendungen fortfahren. - Setzen Sie einen Trichter in eine saubere 50-ml-Röhre in Eis gelegt. Legen Sie eine Schicht Filtrationstuch in den Trichter und pre-wet es mit 5 ml STE. Verwerfen Sie den Durchfluss nicht.
- Gießen Sie das homogenisierte Gewebe in den Trichter. Spülen Sie die Schleifröhre mit 15 ml STE, rekapitulieren und umkehren die Tube, um die Wände und Deckel zu spülen, und gießen Sie in den FunnEl.
- Entfernen Sie vorsichtig die keramischen Steine und drücken Sie dann das Filtrationstuch in den Trichter.
HINWEIS: Handschuhe zwischen den Proben wechseln, um eine Kreuzkontamination zu vermeiden. - Wickeln Sie die Schlauchkappen mit Parafilm ein, um Verschüttungen zu vermeiden. Zentrifugation bei 2.000 xg für 10 min bei 4 ºC.
- Den Überstand sorgfältig mit einer serologischen Pipette absaugen (Vermeiden Sie es, das Pellet zu stören) und legen Sie es in ein 50 mL Hochgeschwindigkeitszentrifugenröhrchen (wenn die Rohre keine festen Dichtungsdichtungen haben, wickeln Sie die Schlauchkappen mit Parafilm ein, um Verschüttungen zu vermeiden). Die Pellets verwerfen.
- Die Röhrchen innerhalb von 0,1 g unter Verwendung von STE ausgleichen und den resultierenden Überstand für 20 min bei 18.000 xg und 4 ºC zentrifugieren. Um die Röhren auszugleichen, legen Sie einen kleinen Becher Eis auf die Waage, tarieren Sie die Waage und wiegen Sie die Proben auf Eis, um sie kalt zu halten. Alternativ verwenden Sie eine Waage und eine Dunstabzugshaube in einem kalten Raum.
- Den Überstand verwerfen. Füge 1 ml ST zum Pellet hinzu und lege dich sanft wieder aufEinen weichen Pinsel. Fügen Sie 24 ml ST (Endvolumen von 25 ml) hinzu und mischen / wirbeln ( dh drücken Sie den Pinsel auf der Seite des Rohres, um alle Flüssigkeit zu entfernen).
- Die Röhrchen innerhalb von 0,1 g mit ST ausgleichen. 20 min bei 18.000 xg und 4 ºC zentrifugieren. Mittlerweile DNaseI-Lösung vorbereiten (siehe Tabelle 1 für Lager- und Arbeitslösungsrezepte). Für jede Probe ein 200 μl Aliquot in einem 1,5 mL Röhrchen herstellen.
- Den Überstand verwerfen, das Röhrchen abtupfen und das Pellet (noch in einem Hochgeschwindigkeits-Zentrifugenröhrchen) in 300 μl ST mit einem weichen Pinsel wieder suspendieren. Legen Sie den Pinsel in das zuvor hergestellte 1,5 ml-Röhrchen mit 200 μl DNaseI-Lösung und drehen Sie den Pinsel, um eventuell vorhandene Restpellets in der Bürste zu entfernen. Die DNaseI-Lösung wieder in das Hochgeschwindigkeits-Zentrifugenröhrchen pipettieren und vorsichtig zum Mischen schwenken.
- Inkubieren bei 37 ° C für 30 min in einem Wasserbad (Ummantelung parafilm um die Oberseite des Rohres, um Kondensationsleck zu verhindernG in die Kappe). Vorsichtig durch zweimaliges Wirbeln während der Inkubation mischen.
- Die Pelletmischung vorsichtig mit einer Pipettenspitze mit einer breiten Öffnung aus dem Röhrchen pipettieren und in ein 1,5 mL-Flachschlauch geben. Fügen Sie 500 μl 400 mM EDTA, pH 8,0, zum Hochgeschwindigkeits-Zentrifugenröhrchen hinzu und putzen Sie vorsichtig, um alle restlichen Pellets aus dem Röhrchen zu bringen. Übertragen Sie das EDTA auf das gleiche 1,5-ml-Röhrchen mit niedriger Bindung als Pellet-Mischung und mischen Sie vorsichtig durch Inversion.
- Zentrifugieren bei 18.000 xg für 20 min bei 4 ºC. Verwerfe den Überstand, löse das Röhrchen und verwende sofort die DNA-Isolierung. Falls erforderlich, gefrieren Pellets bei -20 ºC, aber dies kann zu einer Renditeverringerung führen, da Rest DNaseI die Proben-DNA abbauen kann, wenn sie nicht sofort verarbeitet wird.
- 5 g frisches Gewebe ernten und in kaltem, sterilem Wasser in einem gekühlten Becher auf Eis spülen.
- DNA-Extraktion aus isolierten Organellen unter Verwendung eines kommerziellen Spaltenbasierten Ansatzes
HINWEIS: Siehe das Kit-Handbuch für das vollständige Protokoll 25 und siehe unten für Änderungen. PrDirekt von der organellaren Isolierung zur DNA-Extraktion wird bevorzugt. Wiederholtes Einfrieren und Auftauen reduziert die DNA-Fragmentgrößen und führt zu einem DNA-Abbau durch Rest DNaseI. Begrenzen Sie Vortexen oder kräftiges Pipettieren, da dies die DNA scheren kann. Die Verwendung von niedrigbindenden Mikrozentrifugenröhrchen wird empfohlen, um die DNA-Wiederherstellung zu maximieren.- DNA-Extraktionsverfahren
HINWEIS: Lesen Sie das detaillierte kommerzielle Protokoll 25, bevor Sie beginnen, um sicherzustellen, dass die Puffer ordnungsgemäß hergestellt / gespeichert sind und dass die Spin-Säulen-Verfahren verstanden werden.- Füge 180 μl Puffer ATL direkt in das Röhrchen mit dem Pellet hinzu (aufgetaut, wenn vorher gefroren und auf Raumtemperatur auf der Tischplatte äquilibriert).
- Gehen Sie mit Schritt 3 im Protokoll für "DNA-Reinigung von Geweben" im Kit-Handbuch mit den folgenden Modifikationen vor: Eine 30-minütige Lyse in Schritt 3, schließt die optionale RNase A-Verdauung ein und eluiert in 3 x 200 μl AE ( Jeder in eine sePflanzen und dann die Elutionen kombinieren).
- Sichern Sie ein Aliquot (mindestens 20 μL) für qPCR (siehe Schritt 4.1). Um vor dem Konzentrieren zu quantifizieren, speichern Sie zusätzliche 1 μl für hochempfindliche Quantifizierung.
- Falls gewünscht, die Probenkonzentration vornehmen.
- DNA-Extraktionsverfahren
- Probenkonzentration mit handelsüblichen Filtereinheiten
HINWEIS: Weitere Informationen finden Sie im kommerziellen Protokoll 26 . Abhängig von der nachgeschalteten Verwendung kann es nicht notwendig sein, die Probenkonzentration durchzuführen ( z. B. für Endpunkt-PCR- und qPCR-Anwendungen). Für die NGS-Bibliothekskonstruktion wird es jedoch wahrscheinlich sein, die nach der DNA-Extraktion erhaltene verdünnte organellare DNA zu konzentrieren.- Konzentrationsspaltenverfahren
- Die leere Filtereinheit (ohne Schlauch) auf einem sauberen Stück Wägepapier auf einer digitalen Analysenwaage vorsichtig vorwiegen (siehe Tabelle 2 ). Notieren Sie das Gewicht.
- PiDie kombinierten Elutionen in die Filtereinheit einlegen und sorgfältig wiegen.
HINWEIS: Das Handelshandbuch 26 sagt, dass die maximale Filtermenge der Filtereinheit 500 μl beträgt, aber bis zu 575 μL können dem Gerät sofort ohne Überlauf zugeführt werden. - Setzen Sie die gefüllte Filtereinheit vorsichtig in einen Schlauch (mit den Säulen versehen). Zentrifugation bei 500 xg für die gewünschte Zeit, um das erforderliche Konzentratvolumen zu erreichen. Bei einem Probenvolumen von ~ 575 μl ergibt ein 20-minütiger Spin normalerweise ein Konzentratvolumen von 15 - 30 μl.
- Die Filtereinheit aus dem Rohr nehmen und wieder wiegen. Verwenden Sie die Tabelle, um festzustellen, ob das gewünschte Konzentratvolumen erreicht ist. Wenn nicht, nochmals bei 500 xg für eine kürzere Zeit zentrifugieren und wieder wiegen; Wiederholen, bis das gewünschte Konzentratvolumen erreicht ist.
- Legen Sie eine neue Röhre (mit den Säulen versehen) über die Oberseite der Filtereinheit und invertieren. Zentrifuge für 3 min bei 1.000 xg, um die co zu übertragenIn die Röhre zentrieren
- Bestimmen Sie das gewonnene Volumen. Dies ist in der Regel ~ 3 - 5 μL kleiner als das berechnete Volumen, aufgrund der Filterretention. Wenn überkonzentriert, mit sterilem Wasser oder TE verdünnen, um das gewünschte Volumen zu erreichen.
- Quantifizierung der DNA mit hochempfindlicher Quantifizierung (pro Herstelleranleitung).
- Konzentrationsspaltenverfahren
3. Methode Nr. 2: Methyl-Fraktionierung (MF) Ansatz zur Anreicherung von organellaren DNA aus der gesamten genomischen DNA
HINWEIS: Dieses Protokoll wurde von einem vom Anwender entwickelten Genomic Tip Kit DNA Extraktionsprotokoll für Pflanzen und Pilze 27 und dem kommerziellen Microbiome DNA Enrichment Kit Protokoll 28 modifiziert. In der Theorie kann jedes DNA-Isolationsprotokoll, das hochmolekulare DNA liefert, für das Pulldown verwendet werden. Für die Kurzlesen-Sequenzierung ist jede Extraktion, die überwiegend> 15 kb-Fragmente ergibt, für den Einsatz im Pulldown ausreichend. Für loNg-Lese-Sequenzierung, größere Fragmente können wünschenswert sein. Daher haben wir dieses Protokoll optimiert, um hochmolekulare DNA zu erhalten.
- Isolierung der Gesamt-DNA
HINWEIS: Siehe Liste der benötigten Geräte und Reagenzien und bereiten die erforderlichen Puffer und Arbeitsvorräte für Methode Nr. 2 ( Tabelle 1 ) vor. Fügen Sie Lysing-Enzyme dem Lysepuffer hinzu, um die Lysepuffer-Arbeitslösung zu machen. Schalten Sie den Thermomixer ein und stellen Sie ihn auf 37 ° C. Das Wasserbad auf 50 ° C stellen und QF-Puffer in das Bad geben. Legen Sie 70% EtOH in den Gefrierschrank und stellen Sie die Mikrozentrifuge auf 4 ° C.- Gesamt-DNA-Extraktion unter Verwendung von kommerziellen DNA-Extraktionsspalten
HINWEIS: Bevor Sie beginnen, lesen Sie bitte das Handelshandbuch 29 für detaillierte Informationen über die Verwendung der Schwerkraft-Anionenaustauschsäulen. Die Säulen können mit einem speziellen Rack aufgebaut oder über die mitgelieferten Kunststoffringe über die Rohre gelegt werden. Alle Schritte, einschließlich der gEnomische Spitzen, sollte erlaubt werden, durch Schwerkraftfluss zu gehen, und Restflüssigkeit sollte NICHT durch gezwungen werden.- Mischen Sie 20 mg gefrorenes Gewebe in flüssigem Stickstoff in einem 2-mL-Flachschlauch mit handgehaltenen Schleifstößeln, die für 2-ml-Röhrchen ausgelegt sind.
- Füge 2 ml Lysepuffer-Arbeitslösung hinzu (die Röhrchen werden sehr voll).
- In einem Thermomischer bei 37 ° C für 1 h unter leichtem Rühren bei 300 U / min inkubieren. Ist ein Thermomixer nicht vorhanden, so ist die Inkubation auf einem Heizblock und das Mischen durch sanftes Flackern alle 15 min eine geeignete Alternative.
- Man gibt 4 μl RNase A (100 mg / ml, Endkonzentration von 200 μg / ml) zu. Invertieren, um in einem Thermomixer für 30 min bei 37 ° C zu mischen und zu inkubieren, unter leichtem Rühren bei 300 U / min.
- Füge 80 μl Proteinase K (20 mg / ml, Endkonzentration von 0,8 mg / ml) zu, ummischen und in einem Thermomixer für 2 h bei 50 ° C unter leichtem Rühren bei 300 U / min inkubieren.
- 10 min bei 4 ° C und 1 zentrifugieren5.000 xg, um die unlöslichen Trümmer zu pellet.
- Während die Proben zentrifugieren, die Säulen mit 1 ml Puffer QBT äquilibrieren und die Säule durch Schwerkraftfluss leeren lassen.
- Verwenden Sie eine Pipettenspitze mit breiter Bohrung, um die Probe sofort zu verteilen (das Pellet zu vermeiden) auf die äquilibrierte Säule zu geben und es vollständig durch die Säule zu fließen. Wenn die Probe vor dem Auftragen auf die Säule trübe, filtriert oder zentrifugiert wird (siehe Handelshandbuch für Details 29 ).
- Sobald die Probe vollständig in das Harz eingetreten ist, waschen Sie die Säule mit 4 x 1 ml Puffer QC.
- Die Säule über einem sauberen, 2 mL, niedrig bindenden Mikrozentrifugenröhrchen aufhängen. Eluieren Sie die genomische DNA mit 0,8 ml Puffer QF, der bei 50 ° C vorgewärmt ist.
- Fälligkeit der DNA durch Zugabe von 0,56 ml (0,7 Volumina Elutionspuffer) von Raumtemperatur-Isopropanol zu der eluierten DNA.
- Durch Inversion (10X) mischen und sofort für 20 min bei 15.000 xg und 4 ° C zentrifugieren. PflegeDen Überstand vollständig entfernen, ohne das glasige, lose angebrachte Pellet zu stören.
- Das zentrifugierte DNA-Pellet mit 1 ml kaltem 70% igem Ethanol waschen. 10 min bei 15.000 xg und 4 ° C zentrifugieren.
- Entfernen Sie vorsichtig den Überstand (seien Sie vorsichtig mit diesem Schritt auch), ohne das Pellet zu stören. Luft trocknen für 5-10 min und resuspendieren die DNA in 0,1 ml Elutionspuffer (EB). Die DNA über Nacht bei Raumtemperatur auflösen. Vermeiden Sie Pipettieren, die die DNA scheren können.
- Quantifizieren Sie die Proben mit einem hochempfindlichen DNA-Quantifizierungs-Assay (nach Herstelleranweisungen).
- Gesamt-DNA-Extraktion unter Verwendung von kommerziellen DNA-Extraktionsspalten
- Wulstbasierte Fraktionierung von methylierter und unmethylierter DNA
HINWEIS: Eine neuere Veröffentlichung zeigte die Verwendung eines handelsüblichen Kits 28 , der einen Pulldown-Ansatz nutzt, wobei ein CpG-spezifisches Methyl-bindendes Domänenprotein verwendet wird, das mit dem humanen IgG-Fc-Fragment (MBD2-Fc-Protein) an die Fraktion fusioniert istAß pflanzliche organelläre Genome (unmethyliert) aus Kerngenom (hochmethylierter) Gehalt 23 . Die Fraktionierungseffizienz in Weizenproben wurde bisher nicht mit diesem kommerziellen MF-Kit 28 getestet.- Dinge vor dem Start
- Frisch zubereiten 80% Ethanol (mindestens 800 μl pro Reaktion). 5x binden / waschen Puffer auf Eis auftauen und 5 ml 1x Puffer pro Probe vorbereiten (5X Puffer mit sterilem, nukleasefreiem Wasser verdünnen und während des Protokolls auf Eis aufbewahren).
- Bereiten Sie MBD2-Fc-Protein-gebundene magnetische Perlen vor
- Bereiten Sie die erforderliche Anzahl von Perlen-Sets vor. Skalieren Sie die Reaktionen, um zwischen 1 und 2 μg Gesamteingangs-DNA zu verwenden, wobei 160 - 320 μl Perlen benötigt werden. Beachten Sie, dass die unten aufgeführten Reaktionen für 1 μg Gesamteingangs-DNA sind, so dass sie 160 μl Perlen benötigen. Skalen Sie die Reaktionen nach den Bedürfnissen.
- Mit breiten Bohrungen Tipps, vorsichtig pipettieren die Protein A Magnetic BEad slurry auf und ab, um eine homogene Suspension zu schaffen. Alternativ das Röhrchen der Perlen vorsichtig 15 min bei 4 ° C drehen.
HINWEIS: Die Perlen nicht vortexen. - Fahren Sie mit den Anweisungen nach den Anweisungen des Herstellers fort. 28
- Capture methylierte nukleare DNA
- Für jede einzelne Probe werden 1 & mgr; g Eingangs-DNA zu einem Röhrchen gegeben, das 160 & mgr; l MBD2-Fc-gebundene magnetische Kügelchen enthält.
- Addieren Sie 5x Bindungs- / Waschpuffer, je nach dem Volumen der DNA-Eingangsprobe für eine Endkonzentration von 1x (Volumen von 5x Bindungs- / Waschpuffer, um (μL) = Volumen der Eingangs-DNA (μL) / 4) hinzuzufügen. Pipettieren Sie die Probe ein paar Mal auf und ab, um sie mit einer Pipettenspitze zu bremsen.
- Drehen Sie die Röhrchen bei Raumtemperatur für 15 min. Die Proben mit einer breitbandigen Pipettenspitze vorsichtig pipettieren und die Proben 2-3 mal während der Inkubation auffassen, um das Wulstverklumpen zu verhindern.
HINWEIS: Das Pipettieren und FlickiNg ist entscheidend für eine effiziente Pulldown der methylierten DNA.
- Sammle angereicherte, unmethylierte organellare DNA
- Drehen Sie kurz das Röhrchen, das die DNA und das MBD2-Fc-gebundene magnetische Kügelchen enthält. Legen Sie den Schlauch mindestens 5 min auf eine magnetische Zahnstange, um die Perlen an der Seite des Rohres zu sammeln. Die Lösung sollte klar erscheinen.
- Mit weit gebohrten Spitzen, entfernen Sie sorgfältig den gereinigten Überstand, ohne die Perlen zu stören. Übertragen Sie den Überstand (enthält unmethylierte, organellar angereicherte DNA) in ein sauberes, niedrig bindendes 2-mL-Mikrozentrifugenröhrchen. Bewahren Sie diese Probe bei -20 oder -80 ° C auf oder gehen Sie direkt zur Stufe 3.2.6 zur Reinigung vor.
- Eluierte gefangene nukleare DNA aus den MBD2-Fc-gebundenen magnetischen Kügelchen
- Wenn die Kernfraktion auch erwünscht ist, folgen Sie den Anweisungen des Herstellers 28, um die nukleare DNA aus den MBD2-Fc-gebundenen magnetischen Kügelchen zu eluieren; Reinigen wie in Schritt 3.2.7 beschrieben.
- Perlen-basierte Nukleinsäure-Reinigung
- Stellen Sie sicher, dass die Reinigungsperlen bei Raumtemperatur sind und gründlich gemischt werden. Gehen Sie mit dem Protokoll nach den Anweisungen im MF-Kit-Handbuch vor 28 .
HINWEIS: Die Probe kann nun für den NGS-Bibliotheksbau oder eine andere nachgeschaltete Analyse verwendet werden.
- Stellen Sie sicher, dass die Reinigungsperlen bei Raumtemperatur sind und gründlich gemischt werden. Gehen Sie mit dem Protokoll nach den Anweisungen im MF-Kit-Handbuch vor 28 .
4. Probenquantifizierung und Qualitätskontrolle
- QPCR-Assay zur Beurteilung der organellaren Anreicherung
HINWEIS: Die hier aufgeführten qPCR-Reaktions- und Testparameter wurden für den Einsatz auf einem Roche LightCycler 480 konzipiert und müssen eventuell auf unterschiedliche Geräte und Reagenzien eingestellt werden. Wenn qPCR nicht verfügbar ist, können Endpunkt-PCR und Visualisierung auf einem Agarosegel als qualitatives Maß der Probenreinheit verwendet werden, wobei die gleichen Primer und Bedingungen hier beschrieben werden. Amplicon Größen werden ~ 150 bp für alle Primer Sets. Siehe Tabelle 3 für die Primer-SequenzCes und Paarungen.- QPCR Reaktionsaufbau
- Um eine individuelle 20 μl qPCR-Reaktion einzurichten, pipettieren Sie sorgfältig in eine einzige Vertiefung einer 96-well qPCR-Platte: 10 μl 2x SYBR Green I Master; 2 & mgr; l der 10 & mgr; M Vorwärts- und Rückwärts-Primer-Mischung (für eine Endkonzentration von 0,5 & mgr; M); 2 μl Schablone (im Bereich der Standardkurve); Und 6 & mgr; l steriles, nukleasefreies H 2 O. Um Pipettierfehler zu reduzieren, ist es bevorzugt, eine Mastermischung mit allen Reaktionskomponenten mit Ausnahme der Matrize herzustellen. Fügen Sie die Master-Mix auf die qPCR-Platte und fügen Sie dann die Vorlage von Interesse für jede gut. Es sollten drei technische Replikate für jede Probe durchgeführt werden, um die Auswirkungen von Pipettierfehlern zu minimieren.
HINWEIS: Letztlich wird das Verhältnis von nuklearen zu organellaren Quantifizierungszyklen zwischen den Proben verglichen, so dass geringfügige Konzentrationsunterschiede akzeptabel sind. Allerdings sollten die DNA-Konzentrationen etwa im Bereich von ea liegenAndere. - Die Platte mit einer hochwertigen qPCR-Siegelfolie abdichten. Die Proben vorsichtig vortexen und dabei die Blasenbildung vermeiden. Drehen Sie kurz die Platte für 2 min bei 4 ° C, um die Probe zu sammeln und alle kleinen Blasen zu beseitigen.
- Legen Sie die Platte in die Maschine. Führen Sie das qPCR-Programm nach den unten aufgeführten Richtlinien aus.
- Um eine individuelle 20 μl qPCR-Reaktion einzurichten, pipettieren Sie sorgfältig in eine einzige Vertiefung einer 96-well qPCR-Platte: 10 μl 2x SYBR Green I Master; 2 & mgr; l der 10 & mgr; M Vorwärts- und Rückwärts-Primer-Mischung (für eine Endkonzentration von 0,5 & mgr; M); 2 μl Schablone (im Bereich der Standardkurve); Und 6 & mgr; l steriles, nukleasefreies H 2 O. Um Pipettierfehler zu reduzieren, ist es bevorzugt, eine Mastermischung mit allen Reaktionskomponenten mit Ausnahme der Matrize herzustellen. Fügen Sie die Master-Mix auf die qPCR-Platte und fügen Sie dann die Vorlage von Interesse für jede gut. Es sollten drei technische Replikate für jede Probe durchgeführt werden, um die Auswirkungen von Pipettierfehlern zu minimieren.
- QPCR-Reaktionsparameter
HINWEIS: Dies sind Standardparameter, mit Ausnahme des Glühzyklus der Verstärkungsstufe. Passen Sie diese Einstellung an, um bestimmte Primer aufzunehmen, wenn sich diese von den in diesem Protokoll vorgestellten Primern unterscheiden.- Vorinkubieren bei 95 ° C für 5 min mit einer Rampenzahl von 4,4 ° C / s.
- Führen Sie 45 Amplifikationszyklen von (1) 95 ° C für 10 s mit einer Rampenzahl von 4,4 ° C / s durch; (2) 60 ° C für 20 s, mit einer Rampenzahl von 2,2 ° C / s; Und (3) 72 ° C für 10 s mit einer Rampenzahl von 4,4 ° C / s (während (3) erfasste Daten.
- Verwenden Sie ein optioSchmelzkurvenzyklus von 95 ° C für 5 s mit einer Rampenzahl von 4,4 ° C / s; 65 ° C für 1 min mit einer Rampenzahl von 2,2 ° C / s; Und 97 ° C, mit einem kontinuierlichen Erfassungsmodus.
- Verwenden Sie einen Kühlzyklus von 40 ° C für 30 s mit einer Rampenzahl von 1,5 ° C / s.
- Assay-Parameter
- Wählen Sie die SYBR-Vorlage aus. Überprüfen Sie die Programmparameter in der Schaltfläche Experiment. Sobald die Platte geladen ist, kann der Assay gestartet werden, und die Einstellungen können während des Assays eingestellt werden.
- Zuordnen von Samples mit dem Sample Editor. Wählen Sie Abs Quant als Workflow und bezeichnen Sie die Proben als unbekannt, Standards oder Negativkontrollen. Geben Sie Replikate an und füllen Sie die Beispielnamen des ersten Replikats aus. Fügen Sie Konzentrationen und Einheiten zu den Standards hinzu.
- Einrichten von Teilmengen für die Analyse; Diese werden im Teilmengeneditor zugeordnet.
- Für die Analyse wählen Sie Abs Quant / 2nd Derivative Max aus der Liste "Neue Analyse erstellen".Importieren Sie die extern gespeicherte Standardkurve (falls zutreffend) und dann kalkulieren; Der Bericht enthält die ausgewählten Informationen.
- Um eine genaue absolute Quantifizierung für die Bestimmung der Kopienzahl oder -konzentration durchzuführen, verwenden Sie eine Standardkurve, die für die zu testende Probe repräsentativ ist ( z. B. organellare DNA, die aus den obigen Methoden isoliert wurde). Da die Menge an mitochondrialer DNA, die zur Herstellung einer Standardkurve erforderlich ist, zu hoch ist, um mit einer vernünftigen Menge an Gewebe erreicht zu werden, verwenden Sie keine Kopienzahlberechnungen, die von der Software bereitgestellt werden, sondern stattdessen Kreuzungspunkt (Cp) -Werte untersuchen, um die relative Anreicherung zu bestimmen Von organellaren im Vergleich zu nuklearer DNA in den Proben. Vergleichen Sie diese relativen Mengen mit denen der gesamten genomischen DNA (siehe die repräsentativen Ergebnisse ). Test-Primer-Effizienz bei fünf 1: 10-Verdünnungen der gesamten genomischen DNA aus vollständig lichtgewachsenen, zweiwöchigen Weizensämlingen (repräsentative Wirkungsgrade in thE Legende von Abbildung 2 ).
- QPCR Reaktionsaufbau
- Gepulster Gelelektrophorese (PFGE)
HINWEIS: Dieses Protokoll basiert auf Herstellerrichtlinien, um PFGE durchzuführen, um hochmolekulare DNA zu lösen. Siehe Materialtabelle.- Vorbereitung des Gels und Proben
- Befolgen Sie die Richtlinien für Gel und Probenvorbereitung und passen Sie sie an das verfügbare System an.
- Führen Sie die Parameter aus
- Befolgen Sie die Richtlinien für die Einrichtung des Elektrophorese-Systems und verwenden Sie die folgenden Parameter: Anfangsschaltzeit von 2 s, Endschaltzeit von 13 s, Laufzeit von 15 h und 16 min, V / cm von 6 und eingeschlossenem Winkel von 120 ° .
- Fleck und bild das Gel
- Färben Sie das Gel mit einem Farbstoff der Wahl ( zB Ethidiumbromid oder einer geeigneten Alternative) und Bild mit einem geeigneten Gel-Dokumentationssystem.
- Vorbereitung des Gels und Proben
- Verwenden Sie 1 ng DNA als Eingang für die DNA Library Prep Kit, nach den Anweisungen des Herstellers.
- Barcode und Pool die Samples für die Sequenzierung in einem einzigen Lauf. Führen Sie die Sequenzierung nach Herstellerrichtlinien durch.
HINWEIS: Die Pool- und Sequenzierungsparameter können je nach Art des Interesses, dem gewünschten Deckungsgrad und der Plattform, die zur Sequenzierung der Bibliotheken verwendet wird, verändert werden. Zum Beispiel hat eine HiSeq-Spur wesentlich mehr Leistung als eine MiSeq-Spur, so viele weitere Samples können gemultiplext werden. Sequenz eine kleinere Teilmenge von Proben, um zu bestimmen, ob die Abdeckungsniveaus der organellaren Genome für die nachgeschaltete Analyse ausreichend sind.- Überprüfen Sie die Lesequalität mit FastQC 31 , um das Ausmaß der Trimmung und Filterung zu ermitteln, die für die Daten erforderlich ist.
- Trimmen und filtern Sie die Rohwerte mit Trimmomatic 32 oder einem anderen vergleichbaren Programm. Verwenden Sie folgende Einstellungen: ILLUMINACLIP 2:30:10 (um Adapter zu entfernen), FÜHREN 3, TRAILING 3, SLIDINGWINDOW 4:10 und MINLEN 100.
- (PEBI Referenzsequenz NC_007579.1 33 ), Chloroplast (NCBI Referenzsequenz NC_002762.1 34 ) und nukleare 35 Referenzgenome mit Bowtie2 36 , Mit folgenden Einstellungen: -I 0 -X 800 - lichtempfindlich.
- Konvertieren Sie die sam-Ausrichtungsdateien in bam-Format (samtools) und sortieren Sie die Bam-Dateien. Verwenden Sie die Bam-Dateien, um genomweite Abdeckung und per-base Abdeckung mit Bettwäsche zu berechnen. Visualisieren Sie die Ergebnisse mit der R-Plot-Funktion.
- Dinge vor dem Start
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Die in diesem Manuskript vorgestellten Protokolle beschreiben zwei verschiedene Methoden, um für organellare DNA aus Pflanzengewebe zu bereichern. Die hier vorgestellten Bedingungen spiegeln die Optimierung für Weizengewebe wider. Ein Vergleich der Schlüsselschritte in den Protokollen, der erforderlichen Gewebeeingabe und der DNA-Ausgabe sind in Fig. 1 beschrieben . Die Schritte des DC-Protokolls, das wir getestet haben, folgen ähnlichen Bedingungen wie die zuvor beschriebenen ( Abbildung 1A ). Das geerntete Gewebe muss frisch verarbeitet und einer differentiellen Zentrifugation und / oder Gradienten unterworfen werden, um intakte Organellen zu isolieren. Die nukleare DNA wird eliminiert, bevor die Organellen lysiert werden, und schließlich wird die DNA extrahiert und für nachgeschaltete Anwendungen verwendet. Im Gegensatz dazu kann im MF-Protokoll Pflanzengewebe geerntet und vor Gebrauch gelagert werden, und intakte Organellen sind nicht erforderlich. Stattdessen wird die nukleare und organellare DNA aus der Gesamt-gDNA auf der Basis von Der Methylierungsstatus der DNA. Beide Protokolle ergeben etwa gleiche Mengen an organellarer DNA ( Abbildung 1C ). In Bezug auf die gesamte organellare DNA-Ausgabe relativ zur Gewebe-Inputs ist das MF-Protokoll vorteilhaft, wenn das Gewebe begrenzt ist, da eine kleine Probe aus einer einzigen Pflanze verwendet werden kann und die Pflanze für eine weitere Analyse wachsen gelassen werden kann. Typischerweise sind in DC-Protokollen alle Luftgewebe von vielen Sämlingen erforderlich, und diese Pflanzen werden verworfen. Allerdings kann das DC-Verfahren optimiert werden, um spezifisch für einen Organellen-Typ über das andere zu bereichern, was bei dem MF-Ansatz nicht möglich ist. Es ist erwähnenswert, dass die Gesamtzeit für jedes Protokoll in etwa gleich ist, obwohl es im MF-Ansatz weniger Hands-on-Zeit gibt.
Beide Methoden bereichern für organellare DNA, allerdings mit unterschiedlichen Proportionen von Mitochondrien und Plastidsequenzen:
"> Sehr geringe Mengen an gereinigter organellarer DNA werden aus beiden Methoden (in der Größenordnung von ~ 50 - 100 ng, Abbildung 1C ) erhalten. Zur Beurteilung der Ebenen der organellaren Genomanreicherung und der nuklearen Genomkontamination in DNA, die sowohl aus der DC als auch aus der MF isoliert wurde In diesem Assay wurden die relativen Häufigkeiten von drei Ampullen ( dh nuklearspezifische, ACTIN , mitochondrial-spezifische, NAD3 und Chloroplast-spezifische, PSBB ) wurden in der gesamten genomischen DNA beurteilt und die organellare DNA-Fraktion wurde aus beiden Methoden erhalten ( Abbildung 2 ). Die Quantifizierungszyklus (C q ) -Werte wurden für jede Probe untersucht ( 2A ), und weil das C q als der PCR-Zyklus definiert ist, bei dem die Fluoreszenz von der Zielamplifikation über den Hintergrundfluoreszenzpegel ansteigt, haben C q und die Zielhäufigkeit eine Umgekehrte Beziehung. ImDie DC-Probe, die C q von NAD3 und PSBB sind jeweils ~ 17 und ~ 15 Zyklen früher als ACTIN (was ein C q von ~ 36 hat) (siehe Abbildung 2B für C q -Werte und Anreicherungsniveaus). Dies entspricht theoretischen 167.181- und 47.790-fachen Anreicherungen für NAD3 bzw. PSBB , Relativ zu ACTIN in der DC-Probe ( Abbildung 2B , siehe die Legende von Abbildung 2 für die Berechnung). In der gesamten genomischen DNA-Probe sind die Faltungsanreicherungen für NAD3 und PSBB relativ zu ACTIN nur 158 bzw. 10.701. Es ist nicht überraschend, eine höhere Häufigkeit der organellaren Ampullen im Verhältnis zum nuklearen Amplicon in der gesamten genomischen DNA zu finden, da die organellaren Genome in größeren Kopienzahlen pro Zelle als das Kerngenom 37 existieren und dass die Anzahl der Organellen peR-Zelle kann sich je nach Gewebetyp oder der Entwicklungsstufe 38 , 39 unterscheiden . Insgesamt zeigen die Daten an, dass die DC-Methode bevorzugt für Mitochondrien angereichert, was zu erwarten ist, da die Zentrifugationsgeschwindigkeiten für die selektive Isolierung von Mitochondrien und die Verringerung der Atom- und Chloroplasten-Kontamination optimiert sind.Die unmethylierte Fraktion der MF-Gesamt-gDNA zeigt auch eine beträchtliche Anreicherung sowohl der organellaren Ampullen als auch die beibehaltenen relativen Mengen dieser Targets. Die Faltungsanreicherungen für NAD3 und PSBB bezogen auf ACTIN in der unmethylierten Fraktion betragen 20.551 bzw. 1.703.253 ( Abbildung 2A und 2B ). In der methylierten Fraktion sind die Faltungsanreicherungen für NAD3 und PSBB relativ zu ACTIN 31 bzw. 823 indiDass das MBD2-Fc-Protein im Pulldown der methylierten Kern-DNA hoch effizient ist. Da das Chloroplasten-Amplicon eine höhere Häufigkeit als das mitochondriale Amplicon in der gesamten genomischen DNA (~ 6 C q früher), die methylierte Fraktion (~ 5 C q früher) und die nichtmethylierten Fraktionen (~ 6 C q früher) aufweist, deutet dies darauf hin, dass die Native Häufigkeit dieser Ampullen wird durch MDB2-Pulldown nicht wesentlich verändert. Wir konzentrieren uns hier auf die unmethylierte (organellare) Fraktion aufgrund des Interesses an der Sequenzierung dieser Genome spezifisch. Wenn jedoch das Kerngenom das Hauptinteresse ist, würde MF und die anschließende Sequenzierung der methylierten Fraktion eine viel höhere Kerngenomabdeckung als die gesamte genomische DNA-Sequenzierung aufgrund der Verringerung der organellaren DNA-Kontamination ergeben.
Es ist bemerkenswert, dass, wenn qPCR nicht verfügbar ist, die Endpunkt-PCR (mit den gleichen Primern wie für qPCR) die qualita zur Verfügung stelltBewertung der organellaren Reinheit. In diesem Fall zeigen reine organelläre DNA-Proben eine Amplifikation für die mitochondrialen und plastiden Ampullen, aber keine nachweisbare Amplifikation des nuklearen Amplicons auf dem Agarosegel, während die gesamte genomische DNA eine Amplifikation für alle drei Primersätze zeigt, wie in früheren Studien 11 gezeigt , 12
Organellare DNA, die von beiden Methoden isoliert ist, ist für NGS geeignet:
Getrimmte und gereinigte PE-Sequenzierungsfrequenzen (siehe Schritt 4.3) wurden auf zuvor veröffentlichte orangenorganische Referenzgenome abgebildet, und die Menge der für die Kartierung jeder Probe verwendeten Messungen reichte von ~ 800.000 bis 1.100.000 Liest ( Abbildung 3I ). Ergebnisse von Mapping de novo Illumina Sequenzierung liest auf die verfügbaren Weizen Chloroplast und Mitochondrien Genome sind im Einklang mit der qPCR resUlts, mit der DC-Methode, die DNA liefert, die mehr mit der mitochondrialen DNA angereichert ist ( Abbildung 3A und 3B , ~ 80% und ~ 10% der Lesungen zu den mitochondrialen (mt) bzw. Chloroplasten (cp) Genomen) und dem MF-Verfahren Die DNA, die wahrscheinlich die native Häufigkeit der beiden organellaren Genome widerspiegelt ( Fig. 3A und 3B , ~ 20% und ~ 80% der Liest-Maps zu den mt- und cp-Genomen). Bei beiden Methoden übersteigt die theoretische Abdeckung (siehe die Legende von Abbildung 3 für die Berechnung) bei beiden Weizenorganellgenomen eine 100fache Abdeckung (und reicht bis zu ~ 2.000X Abdeckung für das Chloroplastengenom in der unmethylierten Fraktion aus der MF-Methode) Wenn 12 Bibliotheken gemultiplext werden ( Abbildung 3C und 3D , die in dieser Analyse enthaltenen 6 Bibliotheken wurden mit insgesamt 6 Bibliotheken für eine separate Analyse für insgesamt 12 Bibliotheken zusammengefasstGepoolt in einer einzigen Sequenzspur). Eine genauere Sicht auf die Berichterstattung wurde durch die Untersuchung des Bruchteils des Genoms, das in bestimmten Tiefen abgedeckt wurde, sowie auf der Basis der Deckungsstufen ( Abbildung 3E- 3I ) erreicht. Für die MF-Methode betrug die durchschnittliche pro-Basis-Abdeckung ~ 300 - 450X für das mt-Genom und 4.000 - 5.000X für das cp-Genom. Für die DC-Methode betrug die durchschnittliche pro-Basis-Deckung ~ 900 - 1.300 und ~ 500 - 700X für die mt- und cp-Genome. Allerdings gab es einen kleinen Bruchteil sowohl der mt- als auch der cp-Genome, die eine extrem niedrige oder hohe Abdeckung hatten, und dies wurde in der organellaren DNA beobachtet, die von beiden Methoden abgeleitet wurde ( Abbildung 3I ). Regionen mit überdurchschnittlicher Deckung entsprechen wahrscheinlich den Regionen der Homologie zwischen den organellaren Genomen, und Regionen mit geringer Deckung können auf SNPs oder andere kleine Varianten zwischen den von uns sequenzierten Sorten und den veröffentlichten Referenzen hindeuten. Zur Unterstützung dieser Vorstellung, diese SpikesVon hoher Deckung waren am stärksten ausgeprägt für die aus der MF-Methode abgeleitete mt-DNA ( Fig. 3E und 3I ), wahrscheinlich aufgrund der hohen Bedeckung des cp-Genoms bei diesem Verfahren. Unvermeidlich ist die Abdeckung des cp-Genoms im MF-Verfahren uneinheitlicher als das DC-Verfahren ( Fig. 3G und 3H ), was auf leichte Vorspannungen im MBD2-Fc-Pulldown entlang der cp-DNA zurückzuführen sein könnte. Es sind weitere Experimente erforderlich, um festzustellen, warum dies der Fall ist. Unabhängig davon hatten die mt- und cp-Genome eine relativ gleichmäßige Abdeckung mit beiden Methoden und keine großen Bereiche der fehlenden Abdeckung, was durch die Untersuchung des Anteils der in einer bestimmten Tiefe sequenzierten Genome nachgewiesen werden kann ( Abbildung 3E -3H ). Darüber hinaus werden die Ebenen der Abdeckung für beide Genome als ausreichend für Downstream-Analyse, wie zB Variantenanalyse, betrachtet. Wenn es für die Analyse von seltenen Varianten als notwendig erachtet wird, wodurch die Numbe reduziert wirdR von gepoolten Proben würde eine größere Abdeckung erzielen. Alternativ kann eine weitaus größere Anzahl von Proben auf einer HiSeq-Spur gepoolt werden, während eine noch größere Sequenztiefe erreicht wird, wenn auch bei einem Opfer der Sequenzlänge, da die HiSeq-Bibliotheken derzeit im Gegensatz zu PE300-MiSeq-Bibliotheken auf der PE150-Länge begrenzt sind.
Um die Niveaus der nuklearen Genom-Kontamination anhand eines Mapping-Ansatzes zu untersuchen, wurden PE-Kartierungskategorien untersucht. PE-Lesungen können zu einem Referenzgenom in einer Vielzahl von Konfigurationen abbilden. Wenn liest 1 und 2 auf die Referenz in einer Kopf-zu-Kopf-Mode, mit einem bestimmten "erwarteten" Abstand zwischen den beiden Kameraden (basierend auf der durchschnittlichen Einfügungsgröße der Bibliothek und in der Regel als Eingabeparameter in der Mapping-Software angegeben ), Diese PE-Lesungen sollen "konkordant" abbilden. Im Gegensatz dazu ist die "diskordante" Abbildung die Situation, in der die Mates mit einem weniger oder mehr als erwartetenMit dem Referenzgenom oder der Karte in alternativen Konfigurationen (Kopf-an-Schwanz oder Schwanz-zu-Schwanz). Wenn nur ein Kumpel sich an das Referenzgenom anpasst, dann soll das PE gelesen werden, das weder übereinstimmend noch diskordant dem Referenzgenom zugeordnet ist. In allen drei Lese-Mapping-Kategorien können PE-Lesungen ein oder mehrere Male auf das Referenzgenom ausrichten.
Für die DC- und MF-isolierte organellare DNA war die Lesung des Mitochondrien-Genoms vorwiegend in der übereinstimmenden Einmal-Kategorie ( Abbildung 4A ), während die Lügen dem Chloroplasten-Genom in relativ gleichen Anteilen übereinstimmend einmal und übereinstimmend mehr als ( 4B ), wahrscheinlich aufgrund der großen invertierten Wiederholungen, die im Chloroplastengenom vorhanden sind, und auch auf die extrem hohen Abdeckungsniveaus. Jedoch, weniger PE liest sich auf das nukleare Genom und weitgehend mehr als einmal in einemWeder konkordante noch diskordante Mode ( dh nur ein Kumpel kann abbilden). Diese sind am ehesten die Abgrenzung von "off-target" auf Sequenzen im nuklearen Genom, die homolog zu den organellaren Genomen oder falsch zusammengebauten Regionen sind. Nur eine geringe Menge an Liest (<5%), die dem Kerngenom übereinstimmend zugeordnet ist, zeigt eine geringe Niveaus der nuklearen Genomverunreinigung in der aus der DC- oder MF-Methode isolierten organellaren DNA ( Abbildung 4C ), wie sich auch in den qPCR-Ergebnissen widerspiegelt ( Abb 2A ). Die Kernfraktion nach MBD2-Fc-Pulldown aus chinesischen Frühjahrs-nicht-etiolierten Geweben wurde ebenfalls sequenziert, um zu bestimmen, wie effizient das Pulldown bei der Entfernung von unmethylierter DNA ist. Weniger als 1% der Liest in der nuklearen Fraktion-abgeleiteten Bibliothek, die auf organellare Referenzgenome abgebildet ist, während ~ 45% aller Lügen dem Kerngenom zugeordnet sind ( Abbildung 4 ). Allerdings sind die meisten liest in einer diskordanten Art und Weise, wDie wahrscheinlich das hohe Maß an Fehlmontage und Fragmentierung im Weizen-Kernreferenzgenom widerspiegelt. Unabhängig davon deuten die Ergebnisse darauf hin, dass der MBD2-Fc-Pulldown bei der Entfernung von unmethylierter organellarer DNA aus methylierter Nuklear-DNA hoch effizient ist. Es ist bemerkenswert, dass, weil die organellar angereicherte DNA, die aus diesen Methoden resultiert, eine Mischung von Mitochondrien und Chloroplastensequenzen enthält und weil Sequenzähnlichkeiten, die sich aus dem alten Gentransfer zwischen diesen Organellen ergeben, in ihren Genomen bleiben, die richtige Zuordnung der Liest auf die spezifischen Genome müssen bioinformatisch gelöst werden.
Die Etiolierung des Blattgewebes bedeutet nicht zweifellos Organelle Fülle:
Herkömmlicherweise werden etiolierte Gewebe für die mitochondriale DNA-Isolierung der Pflanze bevorzugt, um den Gehalt an Phenolika und Stärken zu verringern, die mit Extraktio interferieren könnenN oder nachgeschalteten Anwendungen 13 . Um festzustellen, ob organelläre Genomanreicherungsgrade durch Wachstumsbedingungen verändert oder verbessert werden konnten, wurden sowohl etiolierte als auch nicht etiolierte Gewebe dem MF-Protokoll und der Sequenzierung unterworfen. Interessanterweise änderte die Etiolierung nicht merklich den Prozentsatz der Lesungen, die den organellaren Referenzgenomen ( Fig. 3A und 3B ) oder der pro-Basis-Abdeckung ( Abbildung 3I ) im Vergleich zu nicht etiolierten Bedingungen zugeordnet waren. Wir isolierten auch organellare DNA unter Verwendung der Differentialzentrifugation mit sowohl etiolierten als auch nicht-etiolierten Geweben, und es wurde ein geringer Unterschied in der Anreicherung zwischen den verschiedenen Geweben unter Verwendung von qPCR (Daten nicht gezeigt) gefunden. Dies deutet darauf hin, dass mehr physiologisch relevante nicht-etiolierte Gewebe für organelläre Sequenzierungsstudien verwendet werden können, ohne nennenswerte Veränderung der Anreicherung.
Qualitätskontrolle schlägt das vorMF-DNA eignet sich am besten für die Langzeit-Sequenzierung:
Da die Langzeit-Sequenzierung für Forscher besser zugänglich ist, wird die Isolierung von hochmolekularen DNA zunehmend wichtiger. Zur Beurteilung der organellaren DNA, die mit jeder Methode für die Unversehrtheit und Qualität isoliert wurde, wurde PFGE verwendet. Die gesamte genomische DNA wandert typischerweise als diffuser Abstrich in PFGE, und das Molekulargewicht wird durch das Protokoll bestimmt und wie die DNA gelagert und nach der Extraktion behandelt wurde. Die gesamte genomische DNA, die mit genomischen Spitzen isoliert wurde, sollte 50 kb übersteigen, was mit PFGE verifiziert wurde ( Abbildung 5 , Spur 2). Die gesamte genomische DNA aus den genomischen Spitzen wird als Input in das Microbiome Enrichment Kit verwendet, um den Kern aus der organellaren DNA zu fraktionieren. Die nach der Fraktionierung erhaltene Kernfraktion nimmt in ihrer Größe ab, bleibt aber um etwa 50 kb ( Abbildung 5 , Spur 4). Das ist nicht suDa die relativ rauere Handhabung der Kernfraktion als Elution aus MBD2-Fc-gebundenen Perlen eine Wärme- und Proteinase-K-Verdauung erfordert. Aufgrund der begrenzten Masse wurde die organellare Fraktion nicht auf PFGE durchgeführt, aber die anschließende Analyse mit der TapeStation zeigte DNA> 50 kb (Daten nicht gezeigt) an. Die mit differentialer Zentrifugation erhaltene organellare DNA hat eine durchschnittliche Masse von ~ 20 kb, die wahrscheinlich durch das ausgedehnte organellare Isolationsprotokoll und die anschließende säulenbasierte DNA-Extraktion und -Konzentration verursacht wird. Gradienten-basierte organellare Isolierung und alternative DNA-Extraktionsmethoden können größere DNA-Fragmentgrößen beibehalten. Unabhängig davon kann DNA der in diesem Protokoll erhaltenen Größe verwendet werden, um 10- oder 15-kb-Sequenzierungs-Lesevorgänge zu erzeugen, wenn während der Bibliotheksvorbereitung Vorsicht geboten wird.
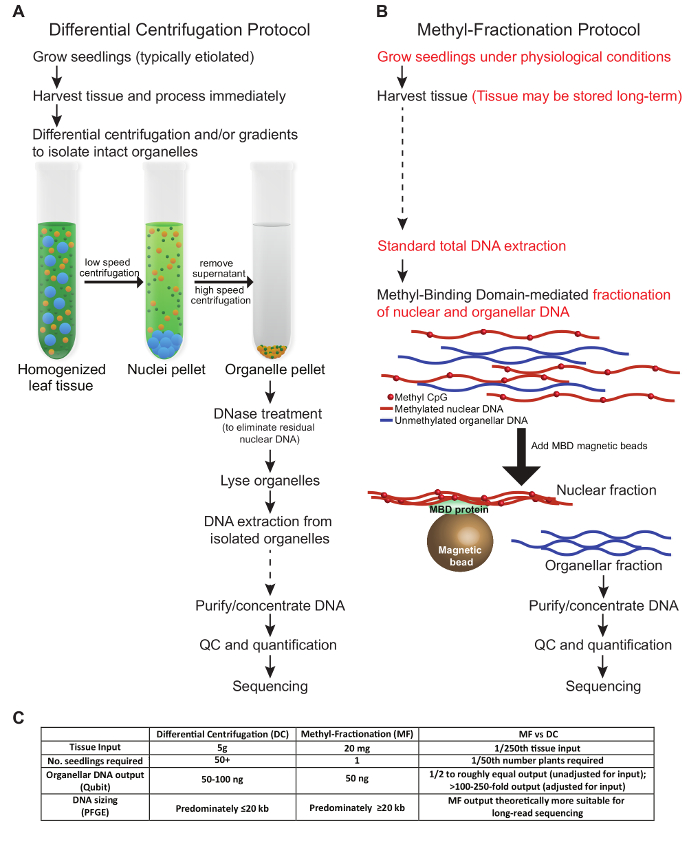
Abbildung 1: Eine vergleichende Ansicht von zwei MethoDs zur Anreicherung für pflanzenorganellare DNA. Ein herkömmliches DC-Protokoll ( A ) steht im Gegensatz zum MF-Protokoll ( B ). Es wird empfohlen, das Einfrieren und Auftauen der Proben zu vermeiden. Jedoch sind die Schritte, bei denen die Proben langfristig gespeichert werden können, mit gestrichelten Pfeilen ( A und B ) angegeben. Wichtige Unterschiede zwischen den Protokollen sind rot markiert ( B ). ( C ) Die Tabelle vergleicht die Methoden in Bezug auf die Gewebeeingabe, die Anzahl der benötigten Pflanzen, die DNA-Ausgabe und die resultierende DNA-Größe. Bitte klicken Sie hier, um eine größere Version dieser Figur zu sehen.

Abbildung 2: Bewertung der nuklearen DNA-Kontamination in der organellaren DNA, die unter Verwendung von zwei Methoden durchgeführt wurde. (
( B ) Die Tabelle zeigt die C- q- Werte, die in der Grafik in ( A ) dargestellt sind, und die Faltungsanreicherung der organellaren Ampullen im Vergleich zu ACTIN . * Fold Anreicherung = 2 (Cq ACTIN - Cq Target) . Die Formel nimmt für jeden Primer-Satz eine perfekte Effizienz von 2 an, da der kleinere AbfallIon jedes Primersatzes von 2 ist vernachlässigbar und hätte wenig Einfluss auf die Berechnung und den Gesamttrend ( ACTIN = 1.961, NAD3 = 1.95 und PSBB = 1.989). Die Primerwirkungsgrade wurden durch eine Standardkurve mit einer Reihe von fünf 1: 10 Verdünnungen der gesamten genomischen DNA ausgewertet. Bitte klicken Sie hier, um eine größere Version dieser Figur zu sehen.

Abbildung 3: Lesen der Kartierung und Theoretische Abdeckung von Chloroplasten und mitochondrialen Genomen. Prozentsatz der Lichter, die den mitochondrialen ( A ) oder Chloroplasten ( B ) chinesischen Frühlingsreferenzgenomen zugeordnet sind. Entsprechende theoretische Abdeckung des chinesischen Frühlings mitochondrialen ( C ) oder Chloroplasten ( D ) ReferenzgenosMes, unter der Annahme, dass Genomgrößen von 450 und 135 kb, berechnet unter Verwendung der gesamten gelesenen Zahlen und des Prozentsatzes der Lese-Mapping auf die verschiedenen Genome. Genomweite Verteilung der Abdeckung für organellare DNA aus der MF-Methode ( E und G ) oder der DC-Methode ( F und H ). Die Daten in den Tafeln E - H stammen aus der chinesischen Frühjahr - etiolierten Probe, aber alle anderen Proben zeigten einen ähnlichen Trend. ( I ) Durchschnittliche, niedrigste und höchste Deckung für alle Proben in den Tafeln A - D. Probenetiketten einschließlich "E" bezeichnen etiolierte Proben und "NE" bezeichnet nicht etiolierte Proben. DC zeigt DNA an, die mit dem Differentialzentrifugationsverfahren isoliert wurde, und Unmethylierte zeigt DNA an, die in der unmethylierten Fraktion nach dem Pulldown mit MBD2-Fc (MF-Protokoll) vorliegt. Proben mit der Bezeichnung "Chris" bezeichnen Weizen Triticum aestivum"Chris" CS bezeichnet Proben von Weizen Triticum aestivum 'Chinese Spring. Anmerkung: Aufgrund der Sequenzhomologie zwischen dem Chloroplasten, den Mitochondrien und den nuklearen Genomen, die sich aus dem alten Gentransfer zwischen den organellaren Genomen sowie zwischen den organellaren und nuklearen Genomen ergeben, kann ein kleiner Prozentsatz der Rohlesungen auf mehrere Genome abbilden. Darüber hinaus sind Liest, die nicht zu entweder organellaren Referenzgenom abbilden, in dieser Figur nicht dargestellt. Daher sind die hier angezeigten Prozentsätze ( A und B ) nicht 100%. Bitte klicken Sie hier, um eine größere Version dieser Figur zu sehen.

Abbildung 4: PE Read Mapping zum Weizen-Kerngenom. Prozentsatz der Kategorien von PE Lesen Sie die Zuordnungstypen zu den mitochondrialen (A) , Chloroplasten (B) oder nuklearen (C) chinesischen Frühlingsreferenzgenomen. - E bezeichnet etiolierte Proben und - NE bezeichnet nicht etiolierte Proben. DC zeigt DNA an, die mit dem Differentialzentrifugationsverfahren isoliert wurde, Unmethyliert zeigt DNA an, die in der unmethylierten Fraktion nach Pulldown mit MBD2-Fc im MF-Protokoll vorliegt, und Methylierung bezeichnet die Kernfraktion nach MBD2-Fc-Pulldown. Proben mit der Bezeichnung "Chris" bezeichnen Weizen Triticum aestivum 'Chris'. CS bezeichnet Proben von Weizen Triticum aestivum 'Chinese Spring'. Nicht gestapelte Lesevorgänge werden nicht angezeigt. Bitte klicken Sie hier, um eine größere Version dieser Figur zu sehen.
Oad / 55528 / 55528fig5.jpg "/>
Abbildung 5: Prüfung der DNA-Qualität mit PFGE. Weizen-Gesamt-Genom-DNA (Spur 2), Weizen-Organell-DNA, erhalten aus Differentialzentrifugation (Spur 3) und die Kernfraktion nach MF mit dem MBD2-Fc-Pulldown-Ansatz (Spur 4) wurden PFGE auf einem 1% Agarosegel mit a 1 kb verlängerte Leiter als Marker (Bahnen 1 und 5) verwendet. Bitte klicken Sie hier, um eine größere Version dieser Figur zu sehen.
| Puffer Name | Rezept | Notizen | Methode |
| STE Puffer | 400 mM Saccharose, 50 mM Tris pH 7,8, 20 mM EDTA pH 8,0, 0,6% (w / v) Polyvinylpyrrolidon (PVP), 0,2% (w / v) Rinderserumalbumin (BSA), 0.1% (v / v) β-Mercaptoethanol (BME) | Puffermischung, die nur Saccharose, Tris und EDTA enthält, kann bis zu einem Monat im Voraus hergestellt und bei 4 ° C gehalten werden. PVP, BSA und BME sollten vor dem Gebrauch frisch zu einem Aliquot der benötigten Puffermenge hinzugefügt werden. | Methode Nr. 1 |
| ST-Puffer | 400 mM Saccharose, 50 mM Tris pH 7,8, 0,6% (w / v) Polyvinylpyrrolidon (PVP), 0,1% (w / v) Rinderserumalbumin (BSA) | Puffermischung, die nur Saccharose und Tris enthält, kann bis zu einem Monat im Voraus hergestellt werden und bei 4 ° C gehalten werden. Beachten Sie, dass der ST-Puffer kein EDTA oder BME enthält und eine niedrigere BSA-Konzentration enthält. | Methode Nr. 1 |
| DNase Lager | 2 mg / ml DNase in 0,15 M NaCl auf eine Stammkonzentration von 2 mg / ml | 200 ul aliquots bei -20 ° C aufbewahren. Zur Vorbereitung der DNase-Arbeitslösung (200 μl DNase-Lösung pro Probe) sieheTabelle 1 unten. Siehe das vollständige Protokoll unten für vollständige Details der DNase-Verdauung. DNase Arbeitslösung sollte frisch hergestellt werden. Um die DNase-Reaktion zu stoppen, ist eine 400 mM EDTA pH 8,0 Lösung erforderlich (Endkonzentration, die benötigt wird, um die Reaktion zu stoppen, beträgt 0,2 M EDTA, siehe vollständiges Protokoll für Details). | Methode Nr. 1 |
| DNase Arbeitslösung | 0,25 mg / ml DNase und 20 mM MgCl 2 in ST-Puffer | Frisch zubereiten, 200 ul pro Probe. Die dargestellten Konzentrationen sind für das Endreaktionsvolumen geeignet, so dass: 62,5 μl 2 mg / ml DNase (bezogen auf das endgültige 500 μl Reaktionsvolumen), 4 μl 1 M MgCl 2 (bezogen auf 200 μl DNase Lösungsvolumen) und 133,5 μl ST Puffer für Ein Endvolumen von 200 μl. | Methode Nr. 1 |
| Lysis Puffer | 20 mM EDTA pH 8,0; 10 mM Tris pH 7,9; 500 mM Guanidin-HCl; 200 mM NaCl; 1% Triton X-100; 0,5 mg / ml lysierende Enzyme ausTrichoderma harzianum | Mischen Sie alle Zutaten außer für lysierende Enzyme und lagern bei Raumtemperatur. Lysing Enzyme sollten frisch zu einem kleinen Aliquot für den sofortigen Gebrauch hinzugefügt werden. | Methode # 2 |
Tabelle 1: Rezepte von hausgemachten Puffern und Arbeitsbeständen.
| Konzentrations-Arbeitsblatt | |||||||
| BEISPIELNAME | Leeres Gerätegewicht (g) | Gewicht des gefüllten Gerätes (g) | Gefülltes Volumen (ul, gefüllt minus leere Gewichte) | Gewicht nach 1. Spin (20 min *, g) | Lautstärke nach dem ersten Spin (ul, gefülltMinus leere Gewichte) | Gewicht nach 2. Spin (X min *, g) | Volumen nach 2. Spin (ul, gefüllt minus leere Gewichte) |
| Beachten Sie, dass das tatsächliche wiederhergestellte Volumen ein paar ul weniger als berechnet Volumen ist. | |||||||
Tabelle 2: Konzentrationsarbeitsblatt.
| Name | Genom-Spezifität | Gensequenz Quelle | Sequenz (5 '- 3') |
| Ta_ACTIN - F. | Nuklear | Gramene Gerüst IWGSC_CSS_1AS_scaff_3272162: 10,663-12,557 | CAGGTATCGCTGACCGTATGA |
| Ta_ACTIN - R | Nuklear | Das gleiche wie oben | GAAGGTAGGGCTGAACAAGAAAC |
| Ta_NAD3 - F. | Mitochondrial | NZBI-Beitritt EU534409.1 | GGTGATGCCAGAAGTCGTTT |
| Ta_NAD3 - R. | Mitochondrial | Das gleiche wie oben | CAGATCAATCTTGTTAGGAGGTACTG |
| Ta_PSBB - F | Chloroplast | NZBI-Beitritt KJ592713.1 | GCTACCTTTGCTTTGCTCTTCT |
| Ta_PSBB - R | Chloroplast | Das gleiche wie oben | GCTGCCTGTTTCCTTGTAGTT |
Tabelle 3: Liste der qPCR-Primer.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Bis heute konzentrieren sich die meisten organellaren Sequenzierungsstudien auf traditionelle DC-Methoden, um für spezifische DNA zu bereichern. Verfahren zur Isolierung von Organellen aus verschiedenen Pflanzen wurden beschrieben, einschließlich Moos 40 ; Monokotten wie Weizen 15 und Hafer 11 ; Und Dicots wie Arabidopsis 11 , Sonnenblumen 17 und Raps 14 . Die meisten Protokolle konzentrieren sich auf das Blattgewebe 13 , 14 , 15 , 16 , 17 , wobei einige für eine Vielzahl von Gewebetypen, einschließlich Samen 11 , angepasst wurden. Die Isolierung von Protoplasten aus Zellorganellen wird auch 41 gezeigt. Dies ist jedoch für alle Systeme nicht zugänglich, noch ist es möglich, wenn das interessierende Gewebe begrenzt ist. Viele dieser OrgaNellar-Isolationsmethoden wurden entwickelt, um intakte Organellen für spezifische Experimente, wie physiologische Studien, zu gewinnen. Diese Protokolle sind mühsam und erfordern typischerweise die Verwendung von Dichtegradienten wie Saccharose- oder Percoll-Gradienten, die bei der Isolierung spezifischer organellarer Fraktionen sehr effizient sind, aber einen großen Gewebeeingang erfordern ( dh über 5 g und über Kilogramm, je nach Der Gewebetyp). Jedoch kann das DC-Verfahren optimiert werden, um spezifische Zellfraktionen, wie die Mitochondrien oder Chloroplasten, durch Ändern von Schleuderdrehzahlen und Dichtegradienten zu bereichern. Im Gegensatz dazu erfordert der MF-Ansatz weit weniger Ausgangsmaterial (20 mg), aber mitochondriale und plastide DNAs werden pro ihrer relativen Häufigkeiten in dem Gewebe vorhanden sein, das für die DNA-Extraktion verwendet wird. Dennoch bietet das MF-Protokoll einen alternativen Ansatz zur Isolierung gemischter organellärer DNA und ist besonders vorteilhaft für den Beginn mit geringen Mengen an Gewebe.
T O beurteilen Sie die Probenreinheit nach der Organellenisolation, die meisten Studien verwenden bisher nur die Endpunkt-PCR und die Gelelektrophorese 11 , 12 . Dies ergibt eine angemessene qualitative Messung der Probenreinheit. Jedoch können niedrige Amplifikationen nicht auf einem Agarosegel sichtbar gemacht werden. Wenige Berichte beinhalten quantitativere Maßnahmen zur Qualitätskontrolle, wie zB qPCR 14 . Für eine quantitative Bewertung der DNA-Probenreinheit, die aus beiden Methoden isoliert wurde, nutzten wir qPCR und Sequenzierung, um zu bestimmen, wieviel nukleare DNA in der Probe verbleibt, sowie die relativen Anteile der mitochondrialen gegenüber der Chloroplasten-DNA. Beide Methoden, die hier ausgewertet werden, sind bei der Entfernung von nuklearer DNA effizient. Beide Methoden ergeben eine Mischung aus mitochondrialer und chloroplastischer DNA, wenn auch in unterschiedlichen Anteilen.
Wachsende Pflanzen im Dunkeln (Etiolierung) sollen dazu beitragen, die organische Isolierung durch eine Reduktion von Phenolen zu erleichternRef "> 13. Allerdings haben wir in diesem Vergleich keinen nennenswerten Vorteil für die Arbeit mit etioliertem Gewebe über lichtgewachsenen Proben gefunden. Obwohl der Anteil der spezialisierten Chloroplasten bei lichtgewachsener Menge höher ist, ist die Gesamtplastidzahl, Spiegelt sich in dem Anteil der Lese-Mapping auf das Chloroplasten-Genom, ist unter unterschiedlichen Lichtverhältnissen unverändert. Für stromabwärtige Funktionsanalysen, wie zB die Beurteilung von Heteroplasmie in verschiedenen Geweben oder unter verschiedenen Stressoren oder für Expressionsanalysen, empfehlen wir die Durchführung einer genomischen Sequenzierung Pflanzen unter physiologisch relevanten Bedingungen gewachsen.
Für die Anwendung mit Short-Read-Sequenzierungstechnologien, verglichen beide Techniken verglichen hier ausreichende DNA-Menge und Qualität. Um jedoch lange Messungen von> 20 kb für Einzelmolekül-Sequenzierungsanwendungen zu erzielen, ist eine größere Menge an DNA mit höherer Qualität erforderlich. Zum Beispiel idealerweise> 1 μg reines orgaNellar-Weizen-DNA mit einem Molekulargewicht> 20 kb ist für in-house, Low-Input-Protokolle für 20-kb Insert-Bibliothekspräparate erforderlich 42 . Neue Benutzer-entwickelte Prototionen mit niedrigem Eingang können die DNA-Anforderungen ( dh auf 50 ng oder sogar weniger 20 ) reduzieren, aber die Herausforderung besteht darin, qualitativ hochwertige, hochmolekulare DNA in die Bibliothekspräparate zu bringen. Es ist wichtig, dass die Mehrheit der DNA> 20 kb beträgt, da kleinere Fragmente bevorzugt in die SMRTbell eingefügt und die Größenverteilung der Bibliothek 43 weggeworfen werden. Wir haben eine Reihe von hausgemachten DNA-Extraktionsprotokollen und eine Reihe von kommerziellen Protokollen für die DNA-Extraktion (nicht gezeigt) versucht. Für Weizenblattgewebe wurde die beste Balance zwischen DNA-Menge und Qualität, insbesondere Länge, unter Verwendung eines kommerziellen Kits 27 , 29 erhalten . Je nach Pflanzenart und Gewebe von Interesse, alternatiDie Extraktionsprotokolle können gleichermaßen geeignet oder fruchtbarer sein. Dennoch schließen wir, dass die Gesamt-Extraktion von hochmolekularen genomischen DNA> 50 kb in der Größe, gefolgt von Fraktionierung mit dem MBD2-Fc-Pulldown-Ansatz 28 , der Langzeit-Sequenzierung aus begrenztem Ausgangsmaterial zugänglich ist. Die zukünftige Arbeit sollte die Grenzwerte des nach der Fraktionierung benötigten Ausgangsmaterials für die Langzeit-Insertionsvorbereitung und die anschließende Langzeitsequenzierung testen. Kritisch könnte dieser Ansatz eine robuste Methode zur Isolierung von DNA aus einer Teilprobe eines einzelnen Blattes bereitstellen, die für eine Langzeitsequenzierung ohne Vollgenom-Amplifikation geeignet ist. Wir gehen davon aus, dass dieser Ansatz leicht an zusätzliche Gewebetypen angepasst werden kann und weitgehend auf andere Pflanzenarten anwendbar ist. Es wird besonders nützlich sein in Situationen, in denen die Gewebemengen begrenzt sind, wie Sequenzierung bei einzelnen Generationen in einem Kreuzungsschema oder in selteneren Gewebetypen.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Die Autoren erklären, dass sie keine konkurrierenden Interessen haben.
Die Erwähnung von Handelsnamen oder kommerziellen Produkten in dieser Publikation dient ausschließlich der Bereitstellung spezifischer Informationen und bedeutet keine Empfehlung oder Anerkennung durch das US Department of Agriculture. USDA ist ein gleichberechtigter Anbieter und Arbeitgeber.
Acknowledgments
Wir möchten die Finanzierung aus dem United States Department of Agriculture-Agricultural Research Service und der National Science Foundation (IOS 1025881 und IOS 1361554) anerkennen. Wir danken R. Caspers für Gewächshauspflege und Pflanzenpflege. Wir danken auch der University of Minnesota Genomics Center, wo die Illumina Bibliothek Vorbereitungen und Sequenzierung durchgeführt wurden. Wir sind auch dankbar für die Kommentare der Zeitschriftenredakteure und vier anonyme Rezensenten, die unser Manuskript weiter gestärkt haben. Wir danken OECD für ein Stipendium für SK, um diese Protokolle für kollaborative Projekte mit Kollegen in Japan zu integrieren.
Materials
| Name | Company | Catalog Number | Comments |
| 2-mercaptoethanol (beta-mercaptoethanol; BME) | Sigma Aldrich | M3148-100ml | |
| 2-propanol (Isopropyl alcohol/isopropanol), bioreagent | Sigma Aldrich | I9516 | |
| agarose, Bio-Rad Cetified Megabase agarose | Bio-Rad | 1613108 | |
| analytical balance | Mettler Toledo | AB54-S | |
| balance | Mettler Toledo | PB1502-S | |
| bovine serum albumin (BSA) | Sigma Aldrich | B4287-25G | |
| Ceramic grinding cylinders, 3/8in x 7/8in | SPEX SamplePrep | 2183 | |
| Cryogenic Blocks compatible with tissue homogenizer for holding 50 mL tubes | SPEX SamplePrep | 2664 | |
| DNaseI | Sigma | DN25 | |
| ethanol, absolute | Decon Laboratories | 2716 | |
| Ethylenediamine Tetraacetic Acid (EDTA), 0.5 M Solution, pH 8.0 | Fisher | BP2482-500 | |
| gel imaging system | |||
| gel stain | Such as GelRed or Ethidium Bromide | ||
| grinding pestle, wide tip for 2 mL conical tubes | |||
| Guanidine-HCl, 8 M solution | ThermoFisher | 24115 | |
| LightCycler 480 SYBR Green I Master | Roche | 4707516001 | |
| liquid nitrogen | |||
| Lysing enzymes from Trichoderma harzianum | Sigma | L1412 | |
| Magnesium Chloride | G Bioscience | 24115 | |
| magnetic rack | ThermoFisher | A13346 | |
| microcentrifuge tubes, LoBind 1.5 mL | Eppendorf | 22431021 | |
| microcentrifuge tubes, standard nuclease-free 1.5 mL | Eppendorf | ||
| microcentrifuge, refrigerated | Sorvall | Legend X1R | Or equivalent product, must be capable of reaching at least 18,000 x g with rotors for 50 mL tubes, Oak Ridge tubes, and 1.5 mL tubes |
| microcentrifuge, room temperature | Eppendorf | 5424 | Or equivalent product, must be capable of reaching at least 18,000 x g with rotor for 1.5 mL and 2 mL microcentrifuge tubes |
| Microcon DNA Fast Flow Centrifugal Filter Units | EMD Millipore | MRCFOR100 | |
| Miracloth, 1 square per sample cut to fit funnel | EMD Millipore | 475855 | |
| NEBNext Microbiome DNA Enrichment Kit | New England Biolabs | E2612L | |
| parafilm | Parafilm M | PM992 | |
| plastic pots and trays | |||
| polyvinylpyrrolidone (PVP) | Fisher | BP431-100 | |
| Proteinase K | Qiagen | 19131 | |
| Pulsed-Field Gel Electrophoresis rig (e.g. CHEF DR III) | Bio-Rad | 1703697 | |
| purification beads, Agencourt AMpureXP beads | Beckman Coulter | A63881 | |
| QIAamp DNA Mini Kit | Qiagen | 51304 | |
| Qiagen 20/g Genomic Tip DNA Extraction Kit | Qiagen | 10223 | |
| Qiagen Buffer EB (elution buffer) | Qiagen | 19086 | |
| Qiagen DNA Extraction Buffer Set | Qiagen | 19060 | |
| QiaRack | Qiagen | 19015 | |
| qPCR machine (e.g. Roche Light Cycler 480) | Roche | ||
| qPCR plate sealing film | Roche | 4729757001 | |
| qPCR plate, 96 well plate | Roche | 4729692001 | |
| Qubit assay tubes | Life Technologies | Q32856 | |
| Qubit Broad Spectrum assay kit | Life Technologies | Q32850 | |
| Qubit High Sensitivity assay kit | Life Technologies | Q32851 | |
| RNaseA | Qiagen | 19101 | |
| Serological pipettes (20 mL) and pipet-aid | Fisher | 13-678-11 | |
| Small funnels, 1 per sample | |||
| Sodium Chloride | Ambion | AM9759 | |
| Soft paintbrush, 2 per sample | |||
| SPEX SamplePrep 2010 Geno/Grinder or another type of tissue homogenizer | SPEX SamplePrep | Or another comparable tissue homogenizer. If you do not have access to a tissue homogenizer, then grinding in a pre-chilled mortar and pestle will suffice (see protocol for details). However, a homogenizer will give more consistent results and total homogenization time is reduced. | |
| Sucrose | Omnipure | 8550 | |
| TBE | |||
| thermomixer | |||
| Tris | Sigma | T2819-100ml | |
| Triton X-100 | Promega | H5142 | |
| tube rotater | |||
| tubes, 50 mL conical polypropylene | Corning | 352070 | |
| tubes, 50 mL high-speed polypropylene | ThermoScientific/Nalgene | 3119-0050 | e.g. Nalgene Oakridge tubes or equivalent |
| vermiculite | |||
| water bath | |||
| water, sterile and certified Nuclease-free | Fisher | 1481 | |
| water, sterile milliQ |
References
- Liberatore, K. L., Dukowic-Schulze, S., Miller, M. E., Chen, C., Kianian, S. F. The role of mitochondria in plant development and stress tolerance. Free Radic Biol Med. 100, 238-256 (2016).
- Samaniego Castruita, J. A., Zepeda Mendoza, M. L., Barnett, R., Wales, N., Gilbert, M. T. Odintifier--A computational method for identifying insertions of organellar origin from modern and ancient high-throughput sequencing data based on haplotype phasing. BMC Bioinformatics. 16 (232), 1-13 (2015).
- Zhang, T., Zhang, X., Hu, S., Yu, J. An efficient procedure for plant organellar genome assembly, based on whole genome data from the 454 GS FLX sequencing platform. Plant Methods. 7 (38), 1-8 (2011).
- Wambugu, P. W., Brozynska, M., Furtado, A., Waters, D. L., Henry, R. J. Relationships of wild and domesticated rices (Oryza AA genome species) based upon whole chloroplast genome sequences. Sci Rep. 5 (13957), 1-9 (2015).
- Iorizzo, M., et al. De novo assembly of the carrot mitochondrial genome using next generation sequencing of whole genomic DNA provides first evidence of DNA transfer into an angiosperm plastid genome. BMC Plant Biol. 12 (61), 1-17 (2012).
- Park, S., et al. Complete sequences of organelle genomes from the medicinal plant Rhazya stricta (Apocynaceae) and contrasting patterns of mitochondrial genome evolution across asterids. BMC Genomics. 15 (405), 1-18 (2014).
- Skippington, E., Barkman, T. J., Rice, D. W., Palmer, J. D. Miniaturized mitogenome of the parasitic plant Viscum scurruloideum is extremely divergent and dynamic and has lost all nad genes. Proc Natl Acad Sci U S A. 112 (27), E3515-E3524 (2015).
- Wicke, S., Schneeweiss, G. M. Chapter 1. Next Generation Sequencing in Plant Systematics. Hörandl, E., Appelhans, M. , Koeltz Scientific Books. (2015).
- Sloan, D. B. One ring to rule them all? Genome sequencing provides new insights into the 'master circle' model of plant mitochondrial DNA structure. New Phytol. 200 (4), 978-985 (2013).
- Woloszynska, M. Heteroplasmy and stoichiometric complexity of plant mitochondrial genomes--though this be madness, yet there's method in't. J Exp Bot. 61 (3), 657-671 (2010).
- Ahmed, Z., Fu, Y. B. An improved method with a wider applicability to isolate plant mitochondria for mtDNA extraction. Plant Methods. 11 (56), 1-11 (2015).
- Ejaz, M., et al. Comparison of small scale methods for the rapid and efficient extraction of mitochondrial DNA from wheat crop suitable for down-stream processes. Genet Mol Res. 13 (4), 10320-10331 (2014).
- Eubel, H., Heazlewood, J. L., Millar, A. H. Isolation and subfractionation of plant mitochondria for proteomic analysis. Methods Mol Biol. 355, 49-62 (2007).
- Hao, W., Fan, S., Hua, W., Wang, H. Effective extraction and assembly methods for simultaneously obtaining plastid and mitochondrial genomes. PLoS One. 9 (9), e108291 (2014).
- Pomeroy, M. K. Studies on the respiratory properties of mitochondria isolated from developing winter wheat seedlings. Plant Physiol. 53 (4), 653-657 (1974).
- Taylor, N. L., Stroher, E., Millar, A. H. Arabidopsis organelle isolation and characterization. Methods Mol Biol. 1062, 551-572 (2014).
- Triboush, S. O., Danilenko, N. G., Davydenko, O. G. A method for isolation of chloroplast DNA and mitochondrial DNA from Sunflower. Plant Mol Biol Rep. 16 (2), 183-189 (1998).
- Pinard, R., et al. Assessment of whole genome amplification-induced bias through high-throughput, massively parallel whole genome sequencing. BMC Genomics. 7 (216), 1-21 (2006).
- Lamble, S., et al. Improved workflows for high throughput library preparation using the transposome-based Nextera system. BMC Biotechnol. 13 (104), 1-10 (2013).
- Raley, C., et al. Preparation of next-generation DNA sequencing libraries from ultra-low amounts of input DNA: Application to single-molecule, real-time (SMRT) sequencing on the Pacific Biosciences RS II. bioRxiv. , (2014).
- Tsai, Y. C., et al. Resolving the Complexity of Human Skin Metagenomes Using Single-Molecule Sequencing. MBio. 7 (1), e01948 (2016).
- Feehery, G. R., et al. A method for selectively enriching microbial DNA from contaminating vertebrate host DNA. PLoS One. 8 (10), e76096 (2013).
- Yigit, E., Hernandez, D. I., Trujillo, J. T., Dimalanta, E., Bailey, C. D. Genome and metagenome sequencing: Using the human methyl-binding domain to partition genomic DNA derived from plant tissues. Appl Plant Sci. 2 (11), 1-6 (2014).
- Noyszewski, A. K., et al. Accelerated evolution of the mitochondrial genome in an alloplasmic line of durum wheat. BMC Genomics. 15 (67), 1-16 (2014).
- Qiagen. QIAamp DNA Mini and Blood Mini Handbook. , 5th ed, Available from: https://www.qiagen.com/ch/resources/ (2016).
- E.M. Corporation. User Guide: Microcon Centrifugal Filter Devices. , Available from: http://www.emdmillipore.com/US/en/product/Microcon-DNA-Fast-Flow-Centrifugal-Filter-Unit-with-Ultracel-membrane,MM_NF-MRCF0R100 (2013).
- Qiagen. User developed protocol: Isolation of genomic DNA from plants and filamentous fungi using the QIAGEN Genomic-tip - (EN). , Available from: https://www.qiagen.com/ch/resources/ (2001).
- New England BioLabs, Inc.. NEBNext Microbiome DNA Enrichment Kit: Instruction Manual Version 4.0. , Available from: http://www.neb.com/~/media/Catalog/All-Products/371BCB5A557C462D95D1E45E15BBFEA3/Datacards or Manuals/E2612Manual.pdf (2015).
- Qiagen. QIAGEN Genomic DNA Handbook. , Available from: https://www.qiagen.com/ch/resources/ (2012).
- PacificBiosciences. Guidelines for Using the BIO-RAD® CHEF Mapper® XA Pulsed Field Electrophoresis System. , Available from: http://www.pacb.com/wp-content/uploads/Unsupported-Guidelines-Using-BIO-RAD-CHEFMapper-XA-Pulsed-Field-Electrophoresis.pdf (2016).
- Andrews, S. FastQC: A quality control tool for high throughput sequence data. , Available from: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (2016).
- Bolger, A. M., Lohse, M., Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 30 (15), 2114-2120 (2014).
- Ogihara, Y., et al. Structural dynamics of cereal mitochondrial genomes as revealed by complete nucleotide sequencing of the wheat mitochondrial genome. Nucleic Acids Res. 33 (19), 6235-6250 (2005).
- Ogihara, Y., et al. Structural features of a wheat plastome as revealed by complete sequencing of chloroplast DNA. Mol Genet Genomics. 266 (5), 740-746 (2002).
- International Wheat Genome Sequencing Consortium (IWGSC). A chromosome-based draft sequence of the hexaploid bread wheat (Triticum aestivum) genome. Science. 345 (6194), (2014).
- Langmead, B., Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat Methods. 9 (4), 357-359 (2012).
- Bendich, A. J. Why do chloroplasts and mitochondria contain so many copies of their genome? Bioessays. 6 (6), 279-282 (1987).
- Kumar, R. A., Oldenburg, D. J., Bendich, A. J. Changes in DNA damage, molecular integrity, and copy number for plastid DNA and mitochondrial DNA during maize development. J Exp Bot. 65 (22), 6425-6439 (2014).
- Ma, J., Li, X. Q. Organellar genome copy number variation and integrity during moderate maturation of roots and leaves of maize seedlings. Curr Genet. 61 (4), 591-600 (2015).
- Lang, E. G., et al. Simultaneous isolation of pure and intact chloroplasts and mitochondria from moss as the basis for sub-cellular proteomics. Plant Cell Rep. 30 (2), 205-215 (2011).
- Tobin, A. K. Subcellular fractionation of plant tissues. Isolation of chloroplasts and mitochondria from leaves. Methods Mol Biol. 59, 57-68 (1996).
- PacificBiosciences. Procedure & Checklist - 10 kb to 20 kb Template Preparation and Sequencing with Low (100 ng) Input DNA. , Available from: http://www.pacb.com/wp-content/uploads/Procedure-Checklist-10-20kb-Template-Preparation-and-Sequencing-with-Low-Input-DNA.pdf (2015).
- PacificBiosciences. Template Preparation and Sequencing Guide. , Available from: http://www.pacb.com/wp-content/uploads/2015/09/Guide-Pacific-Biosciences-Template-Preparation-and-Sequencing.pdf (2014).
