

Summary
Se presentan la comparación y optimización de dos métodos de enriquecimiento de ADN organelar de plantas: centrifugación diferencial tradicional y fraccionamiento del gDNA total basado en el estado de metilación. Evaluamos la cantidad y calidad de ADN resultante, demostramos el rendimiento en secuenciación de próxima generación de lectura corta y discutimos el potencial de uso en la secuenciación de una molécula de larga lectura.
Abstract
Los genomas organelares de las plantas contienen grandes elementos repetitivos que pueden someterse a apareamiento o recombinación para formar estructuras complejas y / o fragmentos sub-genómicos. Los genomas organelas también existen en mezclas dentro de una célula o tipo de tejido dado (heteroplasmia), y una abundancia de subtipos puede cambiar durante el desarrollo o cuando está bajo tensión (desplazamiento subestequiométrico). Se requieren tecnologías de secuenciación de próxima generación (NGS) para obtener una comprensión más profunda de la estructura y la función del genoma organelar. Los estudios de secuenciación tradicionales utilizan varios métodos para obtener ADN organelar: (1) Si se utiliza una gran cantidad de tejido de partida, se homogeneiza y se somete a centrifugación diferencial y / o gradiente de purificación. (2) Si se utiliza una cantidad menor de tejido ( es decir, si las semillas, el material o el espacio son limitados), se realiza el mismo proceso que en (1), seguido por amplificación del genoma completo para obtener suficiente ADN. (3) El análisis bioinformático puede ser usado para buscarEl ADN genómico total y analizar las lecturas organelas. Todos estos métodos tienen desafíos inherentes y compensaciones. En (1), puede ser difícil obtener una cantidad tan grande de tejido de partida; En (2), la amplificación de todo el genoma podría introducir un sesgo de secuenciación; Y en (3), la homología entre los genomas nucleares y organelas podría interferir con el montaje y el análisis. En plantas con genomas nucleares grandes, es ventajoso enriquecer el ADN organelar para reducir los costos de secuenciación y la complejidad de la secuencia para los análisis bioinformáticos. Aquí, comparamos un método de centrifugación diferencial tradicional con un cuarto método, un enfoque adaptado CpG-metil pulldown, para separar el ADN genómico total en fracciones nucleares y organelares. Ambos métodos producen suficiente ADN para NGS, el ADN que está altamente enriquecido para las secuencias organelares, aunque en diferentes proporciones en las mitocondrias y los cloroplastos. Se presenta la optimización de estos métodos para el tejido foliar de trigo y se discuten las principales ventajas ydLas ventajas de cada enfoque en el contexto de la entrada de la muestra, la facilidad del protocolo, y la aplicación descendente.
Introduction
La secuenciación del genoma es una poderosa herramienta para disecar la base genética subyacente de rasgos importantes de las plantas. La mayoría de los estudios de secuenciación del genoma se centran en el contenido del genoma nuclear, ya que la mayoría de los genes se encuentran en el núcleo. Sin embargo, los genomas organelos, incluyendo las mitocondrias (a través de eucariotas) y plastidios (en las plantas, la forma especializada, el cloroplasto, trabaja en fotosíntesis) aportan información genética significativa esencial para el desarrollo de organismos, la respuesta al estrés y la aptitud general 1 . Genomas organelas se incluyen normalmente en el total de extracciones de ADN destinados a la secuenciación del genoma nuclear, aunque los métodos para reducir el número de organelos antes de la extracción de ADN también se emplean [ 2] . Muchos estudios han utilizado resultados de secuenciación de las extracciones totales de gDNA para ensamblar genomas organelos 3 , 4 , 5 ,Xref "> 6 , 7. Sin embargo, cuando el objetivo del estudio es centrarse en los genomas organelares, el uso del gDNA total incrementa los costos de secuenciación debido a que muchas lecturas están" perdidas "en las secuencias de ADN nuclear, particularmente en plantas con genomas nucleares grandes Además, debido a la duplicación y transferencia de secuencias organelares en el genoma nuclear y entre orgánulos, la resolución de la posición correcta de la cartografía de secuenciación lee al genoma adecuado es bioinformatically desafiante 2 , 8. La purificación de los genomas organelares del genoma nuclear es uno Estrategia para reducir estos problemas.Más estrategias bioinformáticas se pueden utilizar para separar las lecturas que mapa a las regiones de homología entre las mitocondrias y los cloroplastos.
Mientras que los genomas organelares de muchas especies de plantas han sido secuenciados, poco se sabe acerca de la amplitud de la diversidad de genomas organelosDisponible en poblaciones silvestres o en piscinas de cría cultivadas. También se sabe que los genomas organelares son moléculas dinámicas que experimentan un reordenamiento estructural significativo debido a la recombinación entre secuencias repetidas 9 . Además, múltiples copias del genoma organelo están contenidas dentro de cada orgánulo, y múltiples organelos están contenidos dentro de cada célula. No todas las copias de estos genomas son idénticos, que se conoce como heteroplasmia. En contraste con el cuadro canónico de "círculos maestros", ahora hay evidencia creciente de una imagen más compleja de las estructuras del genoma organelar, incluidos los círculos sub-genómica, cromosomas lineales, concatamers lineales y estructuras ramificadas [ 10] . El ensamblaje de los genomas organelares de las plantas se complica aún más por sus tamaños relativamente grandes y sus sustanciales repeticiones invertidas y directas.
Los protocolos tradicionales para el aislamiento organelar, la purificación del ADN y el subsiguiente genoma La secuenciación a menudo es engorrosa y requiere grandes volúmenes de entrada de tejido, con varios gramos a más de cientos de gramos de tejido de hoja joven necesarios como punto de partida 11 , 12 , 13 , 14 , 15 , 16 , 17 . Esto hace inaccesible la secuenciación del genoma organelo cuando el tejido es limitado. En algunas situaciones, las cantidades de semillas son limitadas, como cuando es necesario secuenciar sobre una base generacional o en líneas masculinas estériles que tienen que ser mantenidas por cruce. En estas situaciones, el ADN organelar puede purificarse y luego someterse a amplificación de todo el genoma. Sin embargo, la amplificación del genoma completo puede introducir sesgo de secuenciación significativo, que es un problema particular al evaluar la variación estructural, las estructuras sub-genómicas y los niveles de heteroplasmia> 18. Los avances recientes en la preparación de bibliotecas para tecnologías de secuenciación de lectura corta han superado barreras de entrada baja para evitar la amplificación del genoma completo. Por ejemplo, el kit de preparación de biblioteca de Illumina Nextera XT permite que se utilice tan poco como 1 ng de ADN como entrada 19 . Sin embargo, las preparaciones de bibliotecas estándar para aplicaciones de secuenciación de larga lectura, como las tecnologías de secuenciación PacBio o Oxford Nanopore, todavía requieren una cantidad relativamente alta de ADN de entrada, lo que puede representar un reto para la secuenciación del genoma organelo. Recientemente, se han desarrollado nuevos protocolos de secuenciación de larga lectura para reducir las cantidades de entrada y ayudar a facilitar la secuenciación del genoma en muestras en las que es difícil obtener cantidades de microgramos de ADN 20 , 21 . Sin embargo, la obtención de fracciones orgánicas puras de alto peso molecular para alimentar estas preparaciones de biblioteca sigue siendo un desafío.
BuscamosO comparar y optimizar los métodos de enriquecimiento y aislamiento de ADN organelar adecuados para NGS sin necesidad de amplificación de todo el genoma. Específicamente, nuestro objetivo era determinar las mejores prácticas para enriquecer el ADN organelar de alto peso molecular a partir de materiales de partida limitados, como una submuestra de una hoja. Este trabajo presenta un análisis comparativo de los métodos para enriquecer el ADN organelar: (1) un protocolo de centrifugación diferencial tradicional modificado versus (2) un protocolo de fraccionamiento de ADN basado en el uso de un ADN comercialmente disponible CpG-metil-binding domain protein pulldown approach 22 aplicado al tejido vegetal 23 . Recomendamos las mejores prácticas para el aislamiento de ADN organelar de tejido de hoja de trigo, que puede ser fácilmente extendido a otras plantas y tipos de tejidos.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. Generación de materiales vegetales para el aislamiento orgánico y la extracción de ADN
- Crecimiento estándar de plántulas de trigo
- Plante las semillas en vermiculita en pequeñas ollas cuadradas con 4 a 6 semillas por esquina. Traslado a un invernadero o cámara de crecimiento con un ciclo de luz de 16 h, 23 ºC día / 18 ºC noche.
- Regar las plantas cada día. Fertilice las plantas con ¼ de cucharadita de fertilizante granular 20-20-20 NPK durante la germinación y 7 días después de la germinación.
- Etiolación alternativa de las plántulas de trigo
- Siga el paso 1.1, pero coloque las ollas en una cámara de crecimiento oscura, 23 ° C durante 16 h / 18 ºC durante 8 h. Alternativamente, cubra las plantas en el invernadero ( por ejemplo, con un recipiente de almacenamiento, sin embargo, debe mantenerse la ventilación adecuada).
- Crecimiento y recolección de tejidos
- Cultivar las plantas durante 12 - 14 días. Para la mayoría del genotipo del trigoS, 75 - 100 plántulas producen alrededor de 10 - 12 g de tejido, que es suficiente para dos extracciones organelas utilizando el método de centrifugación diferencial (sección 2); Sólo una planta es necesaria si se utiliza la DNA CpG-metilación basada en el enfoque de fraccionamiento organelares de ADN nuclear (sección 3).
- Si se utiliza el método de centrifugación diferencial, recoger el tejido fresco y proceder inmediatamente al procesamiento de las muestras, como se describe en la sección 2.
- Si la utilización del enfoque pulldown CpG-metilo, secciones cosecha 20 mg de tejido de hoja joven en tubos de microcentrífuga (utilizar cualquiera tejido estándar crecido o etiolated, ver los resultados representativos). Congelar a presión sobre nitrógeno líquido y congelar a -80 ºC hasta su uso. Proceda al fraccionamiento del ADN, tal como se describe en la sección 3.
2. Método # 1: Extracción de ADN usando Centrifugación Diferencial (DC)
NOTA: La diferenciaEl protocolo de centrifugación se modificó a partir de dos publicaciones que optimizaron las condiciones para aislar ambos orgánulos pero enriquecer para las mitocondrias 17 , 24 . El protocolo resultante requiere menos tiempo y utiliza menos sustancias químicas tóxicas que los métodos anteriores. Específicamente, hicimos modificaciones a los tampones y pasos de lavado, incluyendo la adición de polivinilpirrolidona (PVP) al tampón de extracción STE y la eliminación del paso de lavado final en tampón NETF, que contiene fluoruro sódico (NaF).
Precaución: La preparación y el uso del tampón STE deben realizarse bajo una campana extractora de humos con equipo de protección personal adecuado, ya que este tampón contiene 2-mercaptoetanol (BME).
- Qué hacer antes de comenzar
- Asegúrese de que todo el equipo esté extremadamente limpio y autoclave cualquier equipo que pueda autoclavarse ( por ejemplo, cilindros deTubos de fuge, etc. ).
NOTA: Se recomiendan puntas de filtro para todos los pasos que requieren pipeteado para evitar la contaminación cruzada. - Vea la lista de equipos y reactivos necesarios y prepare los tampones y las existencias de trabajo requeridos para el Método # 1 ( Tabla 1 ). Enfriar los bloques criogénicos de trituración a -20 ºC y los rotores y tampones a 4 ºC, colocar la microcentrífuga a 4 ºC y activar un baño de agua a 37 ºC.
- Asegúrese de que todo el equipo esté extremadamente limpio y autoclave cualquier equipo que pueda autoclavarse ( por ejemplo, cilindros deTubos de fuge, etc. ).
- Aislamiento de orgánulos
- Cosechar 5 g de tejido fresco y enjuagarlo en agua fría y estéril en un vaso de precipitados refrigerado en hielo.
NOTA: Siempre mantenga las muestras en hielo durante todas las operaciones y transportaciones hacia y desde las centrifugadoras, campanas extractoras de humo, etc. Alternativamente, trabaje en una habitación fría si hay acceso a espacio y equipo suficientes para llevar a cabo el protocolo. - Usando tijeras, corte el tejido de la hoja en pedazos de ~ 1 cm directamente en un tubo de 50 ml que contenga dos rectificado cerámicoCilindros
NOTA: Limpie o cambie las tijeras entre las muestras para evitar la contaminación cruzada. - Si no hay homogeneizador de tejidos, use un mortero y un mazo y siga para reemplazar los pasos 2.2.4 - 2.2.9.
- Cortar el tejido de la hoja en un mortero pre-refrigerado en el hielo. Moler las muestras durante 2 - 3 min en 15 mL de STE (en la campana).
- Vierta el tampón (deje el tejido en el mortero) a través de un embudo que contiene una capa de tela de filtración estéril pre-húmedo (~ 22 a 25 μm de tamaño de poro, vea el protocolo principal para detalles) en otro tubo de 50 ml . Añadir 10 ml adicionales de STE al mortero y al mortero y volver a homogeneizar.
- Verter el tejido homogeneizado y el tampón en el mismo embudo. Enjuague el mortero y el mortero con 10 mL de STE y vierta en el embudo. Apriete y exprima el paño de filtración en el embudo para recuperar la mayor cantidad de líquido posible.
NOTA: Cambie los guantes entre las muestras para evitar la contaminación cruzada. Continuar con el proTocol en el paso 2.2.10.
- Añadir 20 mL de STE (en la campana extractora) a cada tubo de 50 mL.
- Colocar las muestras en bloques de molienda criogénica pre-refrigerados en un aparato de molienda de tejidos y moler las muestras durante 2 x 30 s a 1.750 rpm. Gire las posiciones de la muestra y coloque las muestras en hielo durante ~ 1 min entre las moliendas.
NOTA: En esta etapa se puede usar un mortero y una maja, un mezclador u otro dispositivo de molienda / homogeneización de tejidos. Sin embargo, cada método afectará a la calidad del ADN resultante en diferentes grados, por lo que la longitud y la calidad del ADN deben evaluarse antes de continuar con las aplicaciones aguas abajo. - Inserte un embudo en un tubo limpio de 50 ml colocado en hielo. Coloque una capa de tela de filtración en el embudo y pre-humedezca con 5 mL de STE. No deseche el flujo.
- Verter el tejido homogeneizado en el embudo. Enjuague el tubo de esmerilado con 15 mL de STE, recapaque e invierta el tubo para enjuagar las paredes y la tapa, y verter en el funnEl
- Retire cuidadosamente las piedras de cerámica y luego exprima y exprima el paño de filtración en el embudo.
NOTA: Cambie los guantes entre las muestras para evitar la contaminación cruzada. - Envuelva los tapones del tubo con parafilm para evitar derrames. Centrifugar a 2.000 xg durante 10 min a 4 ºC.
- Aspirar cuidadosamente el sobrenadante con una pipeta serológica (evitar molestar el gránulo) y colocarlo en un tubo de centrífuga de alta velocidad de 50 ml (si los tubos no tienen juntas herméticas, envolver las cápsulas con parafilm para evitar derrames). Deseche los pellets.
- Equilibrar los tubos a 0,1 g usando STE y centrifugar el sobrenadante resultante durante 20 min a 18.000 xg y 4 ºC. Para equilibrar los tubos, coloque un pequeño vaso de hielo sobre la balanza, tarar la balanza y pesar las muestras en hielo para mantenerlas frías. Alternativamente, use una balanza y una campana extractora en una habitación fría.
- Deseche el sobrenadante. Agregue 1 mL de ST a la pella y vuelva a suspender suavementeUn pincel suave. Agregue 24 ml de ST (volumen final de 25 ml) y mezcle / remolino ( es decir, presione el pincel en el lado del tubo para quitar todo el líquido).
- Equilibrar los tubos a 0,1 g usando ST. Centrifugar durante 20 min a 18.000 xg y 4 ºC. Mientras tanto, prepare la solución DNaseI (ver Tabla 1 para las recetas de la solución de trabajo y de trabajo). Para cada muestra, haga una alícuota de 200 μL en un tubo de 1,5 ml.
- Deseche el sobrenadante, borre el tubo y vuelva a suspender el sedimento (todavía en un tubo de centrífuga de alta velocidad) en 300 μl de ST usando un pincel suave. Coloque el pincel en el tubo previamente preparado de 1,5 ml que contenga 200 μL de solución de DNaseI y agite el pincel para eliminar cualquier residuo restante pegado en el cepillo. Pipetar la solución DNaseI de nuevo en el tubo de centrifugación de alta velocidad y suavemente remolinar para mezclar.
- Incubar a 37 ° C durante 30 minutos en un baño de agua (envolver parafilm alrededor de la parte superior del tubo para evitar fugas de condensaciónG en la tapa). Mezclar suavemente por remolino 2 veces durante la incubación.
- Pipetar suavemente la mezcla de pellets fuera del tubo usando una punta de pipeta con un orificio ancho y colocarlo en un tubo de 1,5 ml de bajo enlace. Añadir 500 μL de EDTA 400 mM, pH 8,0, al tubo de centrífuga de alta velocidad y pipetear suavemente para sacar todo el residuo del tubo. Transfiera el EDTA al mismo tubo de 1,5 mL de bajo enlace como la mezcla de pellets y mezcle suavemente por inversión.
- Centrifugar a 18.000 xg durante 20 minutos a 4 ºC. Deseche el sobrenadante, borre el tubo y úselo inmediatamente para aislar el ADN. Si es necesario, congele los gránulos a -20 ºC, pero esto puede resultar en una reducción del rendimiento, ya que la DNasa I residual puede degradar el ADN de la muestra si no se procesa inmediatamente.
- Cosechar 5 g de tejido fresco y enjuagarlo en agua fría y estéril en un vaso de precipitados refrigerado en hielo.
- Extracción de ADN de orgánulos aislados utilizando un enfoque comercial basado en columnas
NOTA: Consulte el manual del kit para el protocolo completo 25 , y consulte las modificaciones a continuación. PrQue procede directamente del aislamiento organelo a la extracción de ADN. La congelación y descongelación repetidas reducirán el tamaño de los fragmentos de ADN y conducirán a la degradación del ADN por la DNasaI residual. Limite el vortex o pipeteado vigoroso, ya que esto puede cortar el ADN. Se recomienda el uso de tubos de microcentrífuga de bajo enlace para maximizar la recuperación del ADN.- Procedimiento de extracción de ADN
NOTA: Antes de comenzar, lea el protocolo comercial detallado 25 para asegurarse de que los búferes están debidamente hechos / almacenados y de que se entienden los procedimientos de columna de giro.- Añadir 180 μl de Tampón ATL directamente en el tubo con el gránulo (descongelado si previamente congelado y equilibrado a temperatura ambiente en el sobre).
- Proceda con el paso 3 en el protocolo para "Purificaciones de ADN de tejidos" en el manual del kit, con las siguientes modificaciones: una lisis de 30 minutos en el paso 3, incluye la digestión opcional de RNasa A y eluye en 3 x 200 μl de AE Cada uno en unoParate y luego combinar las eluciones).
- Guardar una alícuota (al menos 20 μL) para qPCR (ver paso 4.1). Para cuantificar antes de concentrar, ahorre 1 μl adicional para la cuantificación de alta sensibilidad.
- Si se desea, proceda con la concentración de la muestra.
- Procedimiento de extracción de ADN
- Concentración de la muestra con unidades de filtro comerciales
NOTA: Consulte el protocolo comercial 26 para obtener más detalles. Dependiendo del uso en sentido descendente, puede no ser necesario realizar la concentración de la muestra ( por ejemplo, para PCR de punto final y aplicaciones de qPCR). Sin embargo, para la construcción de la biblioteca de NGS, será probablemente necesario concentrar ADN organelar diluido obtenido después de la extracción de ADN.- Procedimiento de la columna de concentración
- Pesar con cuidado (ver Tabla 2 ) la unidad de filtro vacía (sin tubo) sobre un trozo limpio de papel de pesaje sobre una balanza analítica digital. Registre el peso.
- PiPette las eluciones combinadas en la unidad de filtro y pese nuevamente con cuidado.
NOTA: El manual comercial 26 indica que el volumen máximo de la unidad de filtro es de 500 μL, pero puede añadirse hasta 575 μL a la unidad de inmediato sin rebosamiento. - Coloque cuidadosamente la unidad de filtro llena en un tubo (provisto con las columnas). Centrifugar a 500 xg durante el tiempo deseado para lograr el volumen de concentrado requerido. Para un volumen de muestra de ~ 575 μL, un centrifugado de 20 minutos normalmente resultará en un volumen de concentrado de 15-30 μL.
- Retire la unidad de filtro del tubo y vuelva a pesar. Utilice la tabla para determinar si se ha alcanzado el volumen de concentrado deseado. Si no, centrifugar nuevamente a 500 xg durante un período de tiempo más corto y volver a pesar; Repetir hasta que se alcance el volumen de concentrado deseado.
- Coloque un tubo nuevo (proporcionado con las columnas) sobre la parte superior de la unidad de filtro e invierta. Centrifugar durante 3 min a 1.000 xg para transferir el coNcentrate al tubo.
- Determinar el volumen recuperado. Esto suele ser ~ 3 - 5 μL menos que el volumen calculado, debido a la retención del filtro. Si está sobre-concentrado, diluir con agua estéril o TE para lograr el volumen deseado.
- Cuantifique el ADN mediante cuantificación de alta sensibilidad (según las instrucciones del fabricante).
- Procedimiento de la columna de concentración
3. Método # 2: Método de fraccionamiento con metilo (MF) para enriquecer el ADN organelar a partir del ADN genómico total
NOTA: Este protocolo fue modificado a partir de un protocolo de extracción de ADN Genomic Tip Kit desarrollado por el usuario para plantas y hongos 27 y el protocolo comercial Microbiome DNA Enrichment Kit 28 . En teoría, se puede usar cualquier protocolo de aislamiento de ADN que produzca ADN de alto peso molecular para el pulldown. Para secuencias de lectura corta, cualquier extracción que produzca predominantemente fragmentos de más de 15 kb es adecuada para su uso en el pulldown. Por loNg-read, pueden ser deseables fragmentos más grandes. Por lo tanto, optimizamos este protocolo para producir ADN de alto peso molecular.
- Aislamiento del ADN total
NOTA: Consulte la lista de equipos y reactivos necesarios y prepare los tampones y las existencias de trabajo necesarios para el Método # 2 ( Tabla 1 ). Añadir las enzimas de lisis al stock de tampón de lisis para hacer la solución de trabajo del tampón de lisis. Encienda el termomixer y póngalo a 37 ° C. Encienda el baño de agua a 50 ° C y coloque el tampón QF en el baño. Colocar 70% de EtOH en el congelador y poner la microcentrífuga a 4 ° C.- Extracción total de ADN utilizando columnas comerciales de extracción de ADN
NOTA: Antes de comenzar, lea el manual comercial 29 para obtener información detallada sobre el uso de las columnas de intercambio aniónico por flujo de gravedad. Las columnas se pueden montar utilizando un estante especializado o colocadas sobre los tubos utilizando los anillos de plástico suministrados. Todos los pasos, incluyendo el gEnomic tips, se debe permitir que proceda por el flujo de la gravedad, y el líquido residual NO debe ser forzado a través.- Moler 20 mg de tejido congelado en nitrógeno líquido en un tubo de 2 ml de baja unión usando pilas manuales diseñadas para tubos de 2 ml.
- Añadir 2 ml de solución de solución de tampón de lisis (los tubos estarán muy llenos).
- Incubar en un termomixer a 37 ° C durante 1 h con agitación suave a 300 rpm. Si no se dispone de un termomixer, la incubación en un bloque de calor y la mezcla mediante un ligero chasquido cada 15 min son una alternativa adecuada.
- Añadir 4 μl de RNasa A (100 mg / mL, concentración final de 200 μg / mL). Invertir para mezclar e incubar en un termomixer durante 30 minutos a 37 ° C, con agitación suave a 300 rpm.
- Añadir 80 μl de proteinasa K (20 mg / ml, concentración final de 0,8 mg / ml), invertir para mezclar e incubar en un termomixer durante 2 ha 50 ° C, con agitación suave a 300 rpm.
- Centrifugar durante 20 minutos a 4 ° C y 15.000 xg para sedimentar los residuos insolubles.
- Mientras las muestras se centrifugan, equilibrar las columnas con 1 mL de tampón QBT y dejar que la columna se vacíe por flujo por gravedad.
- Utilice una punta de pipeta de diámetro ancho para aplicar rápidamente la muestra (evitar el gránulo) a la columna equilibrada y permitir que fluya completamente a través de la columna. Si la muestra se nubla, vuelva a filtrar o centrifugar antes de aplicarla a la columna (consulte el manual comercial para más detalles 29 ).
- Una vez que la muestra ha entrado completamente en la resina, lavar la columna con 4 x 1 mL de Tampón QC.
- Suspender la columna sobre un tubo de microcentrífuga limpio, de 2 ml, de bajo enlace. Eluir el ADN genómico con 0,8 ml de tampón QF precalentado a 50 ° C.
- Precipitar el ADN añadiendo 0,56 ml (0,7 volúmenes de tampón de elución) de isopropanol a temperatura ambiente al ADN eluido.
- Mezclar por inversión (10X) y centrifugar inmediatamente durante 20 minutos a 15.000 xg y 4 ° C. CuidadoQuite por completo el sobrenadante sin alterar el pellet vítreo y suelto.
- Lavar el sedimento de ADN centrifugado con 1 ml de etanol frío al 70%. Centrifugar durante 10 min a 15.000 xg y 4 ° C.
- Retire cuidadosamente el sobrenadante (tenga cuidado con este paso también) sin alterar el gránulo. Secar al aire durante 5-10 min y resuspender el ADN en 0,1 ml de tampón de elución (EB). Disolver el ADN durante la noche a temperatura ambiente. Evite el pipeteado, que puede cortar el ADN.
- Cuantifique las muestras utilizando un ensayo de cuantificación de ADN de alta sensibilidad (según las instrucciones del fabricante).
- Extracción total de ADN utilizando columnas comerciales de extracción de ADN
- El fraccionamiento a base de perlas de ADN metilado y no metilado
NOTA: Una publicación reciente demostró el uso de un kit disponible comercialmente 28 que aprovecha un enfoque de pulldown utilizando una proteína de dominio de unión a metil CpG específica fusionada al fragmento Fc de IgG humano (proteína MBD2-Fc) a la fracción(No metilados) de genoma nuclear (altamente metilado) contenido 23 . La eficiencia de fraccionamiento en muestras de trigo no se ha probado previamente utilizando este equipo MF comercial 28 .- Qué hacer antes de comenzar
- Preparar de nuevo 80% de etanol (al menos 800 μL por reacción). Colocar 5 veces el tampón de lavado / lavado para descongelar sobre hielo y preparar 5 ml de tampón 1x por muestra (diluir el tampón 5X con agua estéril sin nucleasa y mantenerlo en hielo durante el protocolo).
- Preparar perlas magnéticas unidas a proteínas MBD2-Fc
- Prepare el número requerido de juegos de cuentas. Escala de las reacciones a utilizar entre 1 y 2 μ g de la entrada de ADN total, que requiere 160 - 320 μ l de perlas. Obsérvese que las reacciones enumeradas a continuación son para 1 μg de ADN de entrada total, por lo que requieren 160 μl de perlas. Escala las reacciones según las necesidades.
- Utilizando puntas de gran calibre, pipete suavemente la Proteína A Magnética BEad de la suspensión arriba y abajo para crear una suspensión homogénea. Como alternativa, gire suavemente el tubo de perlas durante 15 min a 4 ° C.
NOTA: No vórtice las perlas. - Proceda con las instrucciones según las instrucciones del fabricante 28 .
- Captura de ADN nuclear metilado
- Para cada muestra individual, añada 1 μg de ADN de entrada a un tubo que contenga 160 μl de perlas magnéticas unidas a MBD2-Fc.
- Se añade 5 x tampón de unión / lavado según sea apropiado dado el volumen de la muestra de entrada de ADN para una concentración final de 1x (volumen de 5x de unión / tampón de lavado para añadir (μL) = volumen de ADN de entrada (μL) / 4). Pipetear la muestra hacia arriba y hacia abajo varias veces para mezclar usando una punta de pipeta de ancho ancho.
- Girar los tubos a temperatura ambiente durante 15 min. Pipetar suavemente las muestras con una punta de pipeta de ancho ancho y sacudir las muestras 2 - 3 veces a lo largo de la incubación para evitar el aglomerado de cuentas.
NOTA: La pipeta y el flickiNg es crítico para garantizar una eficiente desconexión del ADN metilado.
- Recoja el ADN organelar enriquecido, no metilado
- Girar brevemente el tubo que contiene el ADN y la mezcla de perlas magnéticas unidas a MBD2-Fc. Coloque el tubo sobre una rejilla magnética durante al menos 5 minutos para recoger las perlas en el lado del tubo. La solución debe aparecer clara.
- Utilizando puntas de orificio ancho, retire cuidadosamente el sobrenadante despejado sin perturbar las perlas. Transferir el sobrenadante (contiene ADN no metilado, enriquecido en organelos) a un tubo de microcentrífuga de 2 ml limpio y de bajo enlace. Guarde esta muestra a -20 o -80 ° C, o proceda directamente a la etapa 3.2.6 para purificación.
- Eluir el ADN nuclear capturado de las cuentas magnéticas unidas a MBD2-Fc
- Si también se desea la fracción nuclear, siga las instrucciones del fabricante 28 para eluir el ADN nuclear de las perlas magnéticas unidas a MBD2-Fc; Purificar como se describe en el paso 3.2.7.
- Purificación de ácidos nucleicos basada en perlas
- Asegúrese de que las perlas de purificación están a temperatura ambiente y se mezclan a fondo. Proceda con el protocolo según las instrucciones del manual del kit MF 28 .
NOTA: Ahora se puede usar la muestra para la construcción de la biblioteca NGS u otro análisis posterior.
- Asegúrese de que las perlas de purificación están a temperatura ambiente y se mezclan a fondo. Proceda con el protocolo según las instrucciones del manual del kit MF 28 .
- Qué hacer antes de comenzar
4. Cuantificación de la muestra y control de calidad
- Ensayo qPCR para evaluar el enriquecimiento organico
NOTA: La reacción qPCR y los parámetros de ensayo enumerados aquí se diseñaron para su uso en un Roche LightCycler 480 y pueden necesitar ser ajustados para diferentes equipos y reactivos. Si qPCR no está disponible, PCR final y visualización en un gel de agarosa se puede utilizar como una medida cualitativa de la pureza de la muestra, utilizando los mismos cebadores y condiciones descritas aquí. Los tamaños de Amplicon serán ~ 150 pb para todos los conjuntos de cebadores. Véase la Tabla 3 para el secuenciador de cebadorY emparejamientos.- Configuración de la reacción qPCR
- Para configurar una reacción individual de 20 μL de qPCR, pipete cuidadosamente lo siguiente en un solo pocillo de una placa qPCR de 96 pocillos: 10 μl de 2x SYBR Green I Master; 2 μL de la mezcla de cebadores adelante y atrás de 10 μM (para una concentración final de 0,5 μM); 2 μl de plantilla (dentro del intervalo de la curva estándar); Y 6 μl de H 2 O estéril, libre de nucleasa. Para reducir los errores de pipeteado, es preferible hacer una mezcla maestra con todos los componentes de reacción excepto la plantilla. Agregue la mezcla maestra a la placa qPCR y luego agregue la plantilla de interés a cada pozo. Se deben realizar tres repeticiones técnicas para cada muestra para minimizar los efectos del error de pipeteo.
NOTA: En última instancia, la relación entre los ciclos de cuantificación nuclear y organelar se compara entre muestras, por lo que son aceptables pequeñas diferencias en la concentración. Sin embargo, las concentraciones de ADN deberían estar aproximadamente dentro del rango de eaCh otros. - Sellar la placa con una película de sellado qPCR de alta calidad. Suavemente vórtice las muestras, teniendo cuidado de evitar la creación de cualquier burbuja. Gire brevemente la placa hacia abajo durante 2 min a 4 ° C para recoger la muestra y eliminar cualquier burbuja pequeña.
- Cargue la placa en la máquina. Ejecute el programa qPCR según las directrices que se enumeran a continuación.
- Para configurar una reacción individual de 20 μL de qPCR, pipete cuidadosamente lo siguiente en un solo pocillo de una placa qPCR de 96 pocillos: 10 μl de 2x SYBR Green I Master; 2 μL de la mezcla de cebadores adelante y atrás de 10 μM (para una concentración final de 0,5 μM); 2 μl de plantilla (dentro del intervalo de la curva estándar); Y 6 μl de H 2 O estéril, libre de nucleasa. Para reducir los errores de pipeteado, es preferible hacer una mezcla maestra con todos los componentes de reacción excepto la plantilla. Agregue la mezcla maestra a la placa qPCR y luego agregue la plantilla de interés a cada pozo. Se deben realizar tres repeticiones técnicas para cada muestra para minimizar los efectos del error de pipeteo.
- QPCR parámetros de reacción
NOTA: Estos son parámetros predeterminados, excepto para el ciclo de recocido de la etapa de amplificación. Ajuste este ajuste para acomodar cebadores específicos si los utilizados difieren de los cebadores presentados en este protocolo.- Pre-incubar a 95 ° C durante 5 min, con una velocidad de rampa de 4,4 ° C / s.
- Realizar 45 ciclos de amplificación de (1) 95 ° C durante 10 s, con una velocidad de rampa de 4,4 ° C / s; (2) 60ºC durante 20 s, con una velocidad de rampa de 2,2ºC / s; Y (3) 72 ° C durante 10 s, con una velocidad de rampa de 4,4 ° C / s (datos adquiridos durante (3)).
- Utilice una optioCiclo de la curva de fusión nal de 95 ° C durante 5 s, con una velocidad de rampa de 4,4 ° C / s; 65ºC durante 1 min, con una velocidad de rampa de 2,2ºC / s; Y 97 ° C, con un modo de adquisición continua.
- Utilice un ciclo de enfriamiento de 40 ° C durante 30 s, con una velocidad de rampa de 1,5 ° C / s.
- Parámetros del ensayo
- Seleccione la plantilla SYBR. Compruebe los parámetros del programa en el botón Experiment. Una vez cargada la placa, se puede iniciar el ensayo, y los ajustes se pueden ajustar mientras se está ejecutando el ensayo.
- Asigne muestras utilizando el editor de muestras. Seleccione Abs Quant como flujo de trabajo y designe las muestras como desconocidas, estándares o controles negativos. Designe las réplicas y rellene los nombres de muestra de la primera de cada replicación. Añada concentraciones y unidades a las normas.
- Establecer subconjuntos para el análisis; Estos se asignan en el editor de subconjuntos.
- Para el análisis, seleccione Abs Quant / 2nd Derivative Max de la lista "crear nuevo análisis".Importar la curva estándar guardada externamente (si es aplicable) y luego pulsar calcular; El informe contendrá la información seleccionada.
- Para realizar la cuantificación absoluta exacta para la determinación del número o concentración de copias, use una curva estándar que sea representativa de la muestra que se está probando ( por ejemplo, ADN organelar aislado de los métodos anteriores). Dado que la cantidad de ADN mitocondrial requerida para preparar una curva estándar es demasiado alta para ser alcanzada con una cantidad razonable de tejido, no utilice los cálculos de número de copias proporcionados por el software, sino que examine los valores del punto de cruce (Cp) para determinar el enriquecimiento relativo De ADN organelar en comparación con el ADN nuclear en las muestras. Comparar estas cantidades relativas con las del ADN genómico total (ver los resultados representativos ). Pruebe las eficacias del cebador en cinco diluciones 1:10 de ADN genómico total de plántulas de trigo de dos semanas de edad, completamente maduras (las eficiencias representativasLeyenda de la figura 2 ).
- Configuración de la reacción qPCR
- La electroforesis en gel de campo pulsado (PFGE)
NOTA: Este protocolo se basa en las directrices del fabricante para realizar PFGE para resolver el ADN de alto peso molecular. Vea la Tabla de Materiales.- Preparación del gel y muestras
- Siga las pautas para la preparación del gel y de la muestra y adapte al sistema disponible.
- Ejecutar los parámetros
- Siga las directrices para la instalación del sistema de electroforesis y utilice los siguientes parámetros: tiempo de conmutación inicial de 2 s, tiempo de conmutación final de 13 s, tiempo de funcionamiento de 15 h y 16 min, V / cm de 6 y ángulo incluido de 120 ° .
- Mancha e imagen del gel
- Mancha el gel con un colorante de elección ( por ejemplo, bromuro de etidio o una alternativa adecuada) y la imagen con un sistema de documentación de gel adecuado.
- Preparación del gel y muestras
- Utilice 1 ng de ADN como entrada para el kit de preparación de la biblioteca de ADN, de acuerdo con las instrucciones del fabricante.
- Código de barras y agrupe las muestras para la secuenciación en una sola ejecución. Realizar la secuenciación de acuerdo con las directrices del fabricante.
NOTA: Los parámetros de agrupación y secuenciación pueden ser alterados dependiendo de la especie de interés, el nivel de cobertura deseado y la plataforma utilizada para secuenciar las bibliotecas. Por ejemplo, un carril HiSeq tiene una salida sustancialmente mayor que un carril MiSeq, por lo que muchas más muestras pueden ser multiplexadas. Secuencia un subconjunto más pequeño de muestras para determinar si los niveles de cobertura de los genomas organelares son adecuados para el análisis de aguas abajo.- Examine la calidad de lectura con FastQC 31 para determinar el grado de recorte y filtrado requerido para los datos.
- Recortar y filtrar las lecturas sin procesar usando Trimmomatic 32 u otro programa comparable. Utilice los siguientes ajustes: ILLUMINACLIP 2:30:10 (para quitar los adaptadores), LÍDER 3, TRAILING 3, SLIDINGWINDOW 4:10 y MINLEN 100.
- Mapa del extremo emparejado recortada adaptador calidad filtrada y (PE) lee a mitocondrial Resorte chino (NCBI Secuencia de Referencia NC_007579.1 33), cloroplasto (NCBI Secuencia de Referencia NC_002762.1 34), y 35 genomas nucleares de referencia utilizando Bowtie2 36, Con las siguientes configuraciones: -I 0 -X 800 --sensitive.
- Convierta los archivos de alineación sam al formato bam (samtools) y clasifique los archivos bam. Utilice los archivos bam para calcular la cobertura de todo el genoma y la cobertura por base con las herramientas de cama. Visualice los resultados con la función R-plot.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Los protocolos presentados en este manuscrito describen dos métodos distintos para enriquecer el ADN organelar del tejido vegetal. Las condiciones aquí presentadas reflejan la optimización para el tejido de trigo. En la Figura 1 se describe una comparación de los pasos clave en los protocolos, la entrada de tejido requerida y la salida de ADN. Los pasos del protocolo DC que probamos siguen condiciones similares a las descritas anteriormente ( Figura 1A ). El tejido recolectado debe ser procesado recién y sometido a centrifugación diferencial y / o gradientes para aislar orgánulos intactos. El ADN nuclear se elimina antes de que los organelos se lisen, y finalmente, el ADN se extrae y se utiliza para aplicaciones aguas abajo. Por el contrario, en el protocolo MF, el tejido vegetal puede ser cosechado y almacenado antes del uso, y no se requieren orgánulos intactos. En su lugar, el ADN nuclear y organelar se fracciona a partir de gDNA total basado en El estado de metilación del ADN. Ambos protocolos producen cantidades aproximadamente iguales de ADN organelar ( Figura 1C ). En términos de la producción total de ADN organelar en relación con la entrada de tejido, el protocolo MF es ventajoso cuando el tejido está limitado, ya que puede usarse una pequeña muestra de una única planta, y se puede permitir que la planta crezca para un análisis posterior. Típicamente, en los protocolos DC, se requieren todos los tejidos aéreos de muchas plántulas, y estas plantas se descartan. Sin embargo, el método DC se puede optimizar para enriquecer específicamente para un tipo de organelos sobre el otro, lo que no es posible con el enfoque MF. Vale la pena mencionar que el tiempo total para cada protocolo es aproximadamente equivalente, aunque hay menos tiempo práctico en el enfoque MF.
Ambos Métodos Enriquecen para el ADN Organellar, aunque con Proporciones Diferentes de Mitocondrias y Secuestras Plastidicas:
Para evaluar los niveles de enriquecimiento del genoma organelar y la contaminación del genoma nuclear en el ADN aislado de la DC y la MF, se obtuvieron cantidades muy bajas de ADN organelar purificado de cualquiera de los dos métodos (del orden de 50 a 100 ng, Figura 1C ) En este ensayo, la abundancia relativa de tres amplicones ( es decir, nucleares específicos, ACTIN , mitochondrial-específicos, NAD3 , y Cloroplastos específicos, PSBB ) se evaluaron en el ADN genómico total, y la fracción de ADN organelar se obtuvo de ambos métodos ( Figura 2 ]. Los valores del ciclo de cuantificación ( Cq ) se examinaron para cada muestra ( Figura 2A ), y debido a que el Cq se define como el ciclo de PCR en el que la fluorescencia de la amplificación diana aumenta por encima del nivel de fluorescencia de fondo, Cq y la abundancia objetivo tienen una relación inversa. Enla muestra de DC, el C q de NAD3 y PSBB son, respectivamente, ~ 17 y ~ 15 ciclos antes de lo actina (que tiene una q C de ~ 36) (ver Figura 2B para valores de q C y niveles de enriquecimiento). Esto equivale a enriquecimientos teóricos de 167.181 y 47.790 veces para NAD3 y PSBB , respectivamente, Relativo a ACTIN en la muestra DC ( Figura 2B , véase la leyenda de la Figura 2 para el cálculo). En la muestra de ADN genómico total, los enriquecimientos de pliegues para NAD3 y PSBB con respecto a ACTIN son sólo 158 y 10.701, respectivamente. No es sorprendente encontrar una mayor abundancia de los amplicones organelares en relación con el amplicón nuclear en el ADN genómico total, dado que los genomas organelares existen en mayor número de copias por célula que el genoma nuclear 37 y que el número de organelos peR células pueden diferir en función del tipo de tejido o la etapa de desarrollo [ 38 , 39] . En general, los datos indican que el método DC enriquece preferentemente para las mitocondrias, lo cual es de esperar, ya que las velocidades de centrifugación se optimizan para aislar selectivamente mitocondrias y reducir la contaminación nuclear y cloroplástica.La fracción no metilada del gDNA total de MF también muestra un enriquecimiento sustancial de ambos amplicones organelares y se espera que retenga las cantidades relativas nativas de estos dianas. Los enriquecimientos de pliegues para NAD3 y PSBB con respecto a ACTIN en la fracción no metilada son 20,551 y 1,703,253, respectivamente ( Figura 2A y 2B ). En la fracción metilada, los enriquecimientos de pliegues para NAD3 y PSBB con respecto a ACTIN son 31 y 823, respectivamente, indiIndicando que la proteína MBD2-Fc es altamente eficiente en el pulldown del ADN nuclear metilado. Como el amplicón del cloroplasto tiene una mayor abundancia que el amplicón mitocondrial en las muestras de ADN genómico total (~ 6 Cq anterior), la fracción metilada (~ 5Cq anterior) y la fracción no metilada (~ 6Cq anterior), esto sugiere que el La abundancia nativa de estos amplicones no se cambia sustancialmente por MDB2 pulldown. Nos centramos aquí en la fracción unmethylated (organellar) debido al interés en la secuenciación de estos genomas específicamente. Sin embargo, si el genoma nuclear es el interés primario, la MF y la secuenciación subsiguiente de la fracción metilada producirían una cobertura mucho mayor del genoma nuclear que la secuenciación genómica total del ADN, debido a la reducción en la "contaminación" del ADN organelar.
Cabe señalar que si qPCR no está disponible, punto final PCR (utilizando los mismos primers que para qPCR) proporciona la qualitaEvaluación de la pureza organometálica. En este caso, las muestras de ADN orgánico puro mostrarán amplificación para los amplicones mitocondriales y plástidos, pero no se detectó amplificación del amplicón nuclear en el gel de agarosa, mientras que el ADN genómico total muestra amplificación para los tres conjuntos de cebadores, como se demostró en estudios previos 11 , 12 .
El ADN organelar aislado de ambos métodos es adecuado para NGS:
Las lecturas de secuencias de PE recolectadas y limpias (véase el paso 4.3) se asignaron a genomas de referencia de organelos de trigo previamente publicados y la cantidad de lecturas utilizadas para cartografiar cada muestra varió de ~ 800.000 a 1.100.000 lecturas ( Figura 3I ). Resultados de la cartografía de novo Illumina secuenciación lee a la disponible trigo cloroplasto y mitocondrias genomas son compatibles con el qPCR res( Figura 3A y 3B , ~ 80% y ~ 10% del mapa de lecturas al genoma mitocondrial (mt) y al cloroplasto (cp), respectivamente) y el método MF Proporcionando ADN que probablemente refleja la abundancia nativa de los dos genomas organelares ( Figuras 3A y 3B , ~ 20% y ~ 80% de los lee mapas de los genomas mt y cp, respectivamente). En ambos métodos, la cobertura teórica (véase la leyenda de la Figura 3 para el cálculo) de ambos genomas organellar trigo excede cobertura 100X (y oscila hasta cobertura ~ 2,000x para el genoma del cloroplasto en la fracción no metilada del método MF), incluso Cuando 12 bibliotecas están multiplexadas ( Figura 3C y 3D , las 6 bibliotecas incluidas en este análisis se agruparon con 6 bibliotecas adicionales para un análisis separado, para un total de 12 bibliotecasAgrupados en un solo carril de secuenciación). Se obtuvo una visión más detallada de la cobertura examinando la fracción del genoma cubierto a profundidades específicas, así como a los niveles de cobertura por base ( Figura 3E -3I ). Para el método MF, la cobertura media por base fue ~ 300 - 450X para el genoma mt y 4.000 - 5.000X para el genoma cp. Para el método DC, la cobertura promedio por base fue de ~ 900 - 1,300 y ~ 500 - 700X para los genomas mt y cp, respectivamente. Sin embargo, hubo una pequeña fracción de los genomas mt y cp que tenían una cobertura extremadamente baja o alta, y esto se vio en el ADN organelar derivado de cualquiera de los métodos ( Figura 3I ). Las regiones de cobertura superior a la media probablemente corresponden a regiones de homología entre los genomas organelos y regiones con baja cobertura pueden indicar SNP u otras variantes pequeñas entre los cultivares que secuenciamos y las referencias publicadas. En apoyo de esta noción, estos picosDe alta cobertura fueron más pronunciadas para el ADN mt derivado del método MF ( Figuras 3E y 3I ), probablemente debido a la alta cobertura del genoma cp en este método. Inexplicablemente, la cobertura del genoma cp es más desigual en el método MF que el método DC ( Figura 3G y 3H ), lo que podría deberse a ligeros sesgos en el MBD2-Fc desplegable a lo largo del ADN cp. Se necesitarán más experimentos para determinar por qué es este el caso. A pesar de ello, los genomas mt y cp presentaron una cobertura relativamente uniforme con ambos métodos y sin grandes áreas de cobertura faltante, lo que se puede demostrar mediante el examen de la fracción de genomas secuenciados a una profundidad dada ( Figura 3E -3H ). Además, los niveles de cobertura de ambos genomas se consideran suficientes para el análisis de aguas abajo, como el análisis de variantes. Si se considera necesario para el análisis de variantes raras, la reducción del númeroR de muestras agrupadas lograrían una mayor cobertura. Alternativamente, se puede agrupar un número mucho mayor de muestras en un carril HiSeq, al tiempo que se alcanza una profundidad de secuenciación aún mayor, aunque con un sacrificio a la longitud de la secuencia, ya que las bibliotecas HiSeq están actualmente limitadas a la longitud del PE150 en contraste con las bibliotecas PE300 MiSeq.
Para examinar los niveles de contaminación del genoma nuclear mediante un enfoque de cartografía, se examinaron las categorías de mapeo de PE. PE lecturas pueden asignar a un genoma de referencia en una variedad de configuraciones. Cuando las lecturas 1 y 2 se alinean a la referencia de una manera cabeza a cabeza, con una cierta distancia "esperada" entre los dos compañeros (basada en el tamaño medio de inserto de la biblioteca y típicamente especificada como parámetro de entrada en el software de mapeo ), Se dice que estas lecturas de PE mapean "concordantemente". Por el contrario, la cartografía "discordante" es la situación en la que los compañeros se mapean con un menor o mayor de lo esperadoTancia al genoma o mapa de referencia en configuraciones alternativas (de cabeza a cola o de cola a cola). Si sólo un compañero se alinea con el genoma de referencia, entonces se dice que la lectura de PE no se correlaciona ni concordante ni discordantemente con el genoma de referencia. En las tres categorías de lectura, las lecturas de PE pueden alinearse con el genoma de referencia una o varias veces.
Tanto para el ADN organelar DC y MF aislados, el mapeo de lectura al genoma mitocondrial estaba predominantemente en la categoría alineada concordantemente una vez ( Figura 4A ), mientras que las lecturas se correlacionaron con el genoma del cloroplasto en proporciones relativamente iguales de concordancia una vez y concordantemente más de Una vez ( Figura 4B ), probablemente debido a las grandes repeticiones invertidas presentes en el genoma del cloroplasto y también a los niveles de cobertura extremadamente altos. Sin embargo, menos lecturas de PE son mapeadas al genoma nuclear y en gran parte mapeadas más de una vez en unNi concordante ni discordante ( es decir, sólo un compañero es capaz de mapear). Estos son más probable cartografía "fuera de destino" a las secuencias en el genoma nuclear, que son homólogos a los genomas organellar o misassembled regiones. Sólo una cantidad menor de lee (<5%) mapearon al genoma nuclear concordantemente, lo que indica bajos niveles de contaminación genoma nuclear en el ADN organellar aislado del método de DC o MF (Figura 4C), como también se refleja en los resultados de qPCR (Figura 2A ). La fracción nuclear después de MBD2-Fc pulldown de los tejidos no etiolated Primavera Chino también se secuenció para determinar la eficiencia de la pulldown es en la eliminación de ADN no metilado. Menos del 1% de las lecturas de la biblioteca derivada de la fracción nuclear se correlacionan con genomas de referencia organelos, mientras que el ~ 45% de todas las lecturas se correlacionan con el genoma nuclear ( Figura 4 ). Sin embargo, la mayoría lee mapas de una manera discordante, wLo que probablemente refleja los altos niveles de mal montaje y fragmentación en el genoma nuclear nuclear de referencia. Sin embargo, los resultados sugieren que el MBD2-Fc pulldown es altamente eficiente en la eliminación de ADN organelar no metilado de ADN nuclear metilado. Vale la pena señalar que, debido a que el ADN enriquecido en organelos resultante de estos métodos contiene una mezcla de mitocondrias y secuencias de cloroplasto, y debido a que las similitudes de secuencia resultantes de la transferencia de genes antiguos entre estos orgánulos permanecen en sus genomas, la asignación apropiada de lecturas a los específicos Los genomas deben ser resueltos bioinformáticamente.
La etiolación del tejido foliar no se altera apreciablemente Organelle Abundancias:
Tradicionalmente, los tejidos etiolados son preferidos para el aislamiento del ADN mitocondrial de plantas con el fin de disminuir los niveles de fenoles y almidones, que pueden interferir con la extracciónN o aguas abajo 13 . Para determinar si los niveles de enriquecimiento del genoma organelar podrían ser alterados o mejorados por las condiciones de crecimiento, ambos tejidos etiolados y no etiolados fueron sometidos al protocolo MF y secuenciación. Curiosamente, la etiolación no cambió apreciablemente el porcentaje de lecturas que se asignaron a los genomas de referencia organelas ( Figuras 3A y 3B ) o la cobertura por base ( Figura 3I ) en comparación con las condiciones no etioladas. También se aisló el ADN organelar utilizando centrifugación diferencial, tanto con tejidos etiolados y no etiolated, y se encontró poca diferencia en el enriquecimiento entre los diferentes tejidos utilizando qPCR (datos no presentados). Esto sugiere que pueden utilizarse tejidos no etiolados más fisiológicamente relevantes para estudios de secuenciación organelar, sin cambio apreciable de enriquecimiento.
El control de calidad sugiere queEl ADN MF es el más adecuado para la secuenciación de larga lectura:
A medida que la secuenciación de lectura larga se vuelve más accesible para los investigadores, el aislamiento de ADN de alto peso molecular es cada vez más importante. Para evaluar el ADN organelar aislado con cualquiera de los métodos para el intacto y la calidad, se empleó PFGE. El ADN genómico total migra habitualmente como un frotis difuso en PFGE, y el peso molecular está determinado por el protocolo y cómo se almacenó y manipuló el ADN después de la extracción. El ADN genómico total aislado con puntas genómicas debe superar los 50 kb, que se verificó utilizando PFGE ( Figura 5 , carril 2). El ADN genómico total de las puntas genómicas se utiliza como la entrada en el Microbiome Enrichment Kit para fraccionar la nuclear de DNA organelar. La fracción nuclear obtenida después del fraccionamiento disminuye en tamaño, pero permanece centrada alrededor de 50 kb ( Figura 5 , carril 4). Esto no es suDado que el manejo relativamente más áspero de la fracción nuclear como elución de perlas unidas a MBD2-Fc requiere la digestión con calor y proteinasa K. Debido a la masa limitada, la fracción organelar no se hizo funcionar con PFGE, pero un análisis posterior con la TapeStation indicó ADN> 50 kb (datos no mostrados). El ADN organelar obtenido con centrifugación diferencial tiene una masa media de ~ 20 kb, probablemente causada por el protocolo de aislamiento orgánico extendido y la posterior extracción y concentración de ADN a base de columna. El aislamiento organelo basado en gradiente y los métodos alternativos de extracción de ADN pueden mantener tamaños de fragmentos de ADN mayores. Independientemente, el ADN del tamaño obtenido en este protocolo puede usarse para generar lecturas de secuenciación de 10 ó 15 kb si se toma cuidado durante la preparación de la biblioteca.
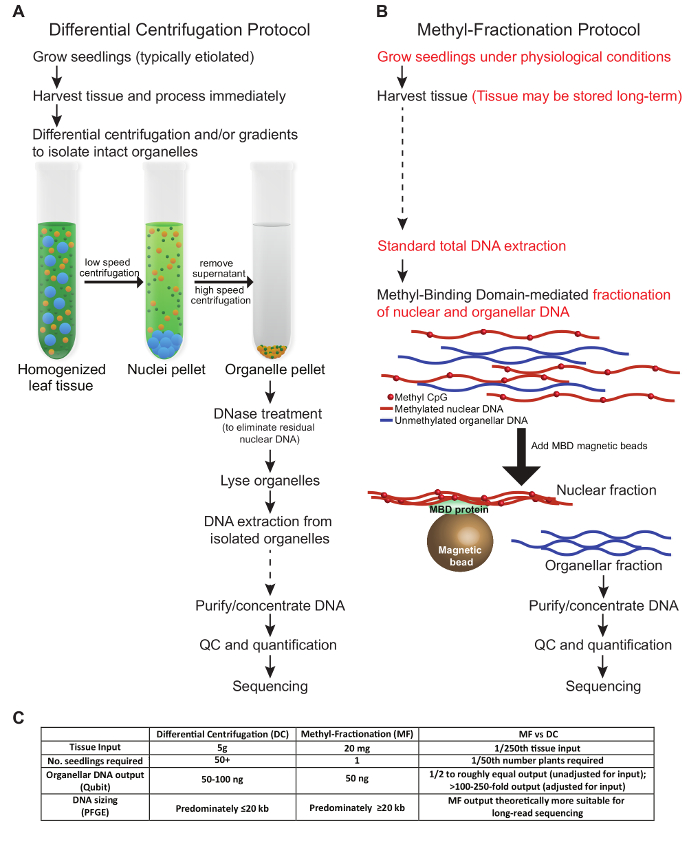
Figura 1: Una vista comparativa de dos methoDs para enriquecer para el ADN organelo vegetal. Un protocolo DC tradicional ( A ) se contrasta con el protocolo MF ( B ). Se recomienda evitar congelar y descongelar las muestras; Sin embargo, las etapas en las que las muestras pueden almacenarse a largo plazo se indican con flechas discontinuas ( A y B ). Las principales diferencias entre los protocolos se destacan en rojo ( B ). ( C ) La tabla compara los métodos en términos de entrada de tejido, número de plantas requeridas, producción de ADN y tamaño de ADN resultante. Haga clic aquí para ver una versión más grande de esta figura.

Figura 2: Evaluación de la contaminación por ADN nuclear en ADN organelo aislado usando dos métodos. (
( B ) La tabla muestra los valores de C q , que se muestran en el gráfico en ( A ), y el enriquecimiento de veces de los amplicones organelares con respecto a ACTIN . * Enriquecimiento de doblado = 2 (Cq ACTIN - Cq Target) . La fórmula supone una eficiencia perfecta de 2 para cada conjunto de cebadores, ya que el deviat menorIon de cada conjunto de cebadores de 2 es insignificante y tendría poco efecto sobre el cálculo y la tendencia general ( ACTIN = 1.961, NAD3 = 1.95 y PSBB = 1.989). Las eficacias del cebador se evaluaron haciendo una curva estándar con una serie de cinco diluciones 1:10 del ADN genómico total. Haga clic aquí para ver una versión más grande de esta figura.

Figura 3: Cartografía de lectura y cobertura teórica de los genomas cloroplastos y mitocondriales. Porcentaje de lecturas mapeadas a los genomas mitocondriales ( A ) o cloroplasto ( B ) de primavera china. Correspondientes cobertura teórica de la Primavera Chino mitochondrial ( C ) o cloroplasto ( D ) referencia genoMes, suponiendo tamaños de genoma de 450 y 135 kb, respectivamente, calculado utilizando el total de los números de lectura y el porcentaje de lectura de la cartografía a los diferentes genomas. Distribución de la cobertura de ADN organelar en todo el genoma a partir del método MF ( E y G ) o del método DC ( F y H ). Los datos de los paneles E - H provienen de la muestra Etiolated Chinese Spring, pero todas las demás muestras mostraron una tendencia similar. ( I ) Cobertura media, mínima y máxima por base para todas las muestras en los paneles A - D. Las etiquetas de muestra incluyendo "E" designan muestras etioladas, y "NE" designa muestras no etioladas. DC indica ADN aislado con el método de centrifugación diferencial y Unmethylated indica ADN que está en la fracción no metilada después de pulldown con MBD2-Fc (protocolo MF). Muestras etiquetadas como "Chris" designan trigo Triticum aestivumChris. CS designa muestras de trigo Triticum aestivum 'Chinese Spring. Nota: Debido a la homología de secuencia entre el cloroplasto, las mitocondrias y los genomas nucleares resultantes de la transferencia de genes antiguos entre los genomas organelares y entre los genomas organelares y nucleares, un pequeño porcentaje de lecturas en bruto puede correlacionarse con múltiples genomas. Además, las lecturas que no se corresponden con el genoma de referencia organelar no se representan en esta figura. Por lo tanto, los porcentajes que se muestran aquí ( A y B ) no son del 100%. Haga clic aquí para ver una versión más grande de esta figura.

Figura 4: Mapeo de lectura de la PE al genoma nuclear del trigo. Porcentaje de categorías de PE Leer los tipos de cartografía a los mitocondriales (A) , cloroplasto (B) , o nucleares (C) primavera chino genomas de referencia. - E designa muestras etioladas y - NE designa muestras no etioladas. DC indica ADN aislado con el método de centrifugación diferencial, Unmethylated indica ADN que está en la fracción no metilada después de pulldown con MBD2-Fc en el protocolo MF, y metilado designa la fracción nuclear después de MBD2-Fc pulldown. Las muestras etiquetadas "Chris" designan trigo Triticum aestivum 'Chris.' CS designa muestras de trigo Triticum aestivum 'Chinese Spring'. Las lecturas sin asignar no se muestran. Haga clic aquí para ver una versión más grande de esta figura.
Oad / 55528 / 55528fig5.jpg "/>
Figura 5: Examen de la calidad del ADN utilizando PFGE. El ADN genómico total de trigo (carril 2), el ADN organelar de trigo obtenido a partir de la centrifugación diferencial (carril 3) y la fracción nuclear después de MF con el procedimiento de pulldown MBD2-Fc (carril 4) se sometieron a PFGE en un gel de agarosa al 1% Escala extendida de 1 kb utilizada como marcador (carriles 1 y 5). Haga clic aquí para ver una versión más grande de esta figura.
| Nombre del búfer | Receta | Notas | Método |
| STE Buffer | Sacarosa 400 mM, Tris 50 mM pH 7,8, EDTA 20 mM pH 8,0, polivinilpirrolidona al 0,6% (p / v), albúmina de suero bovino al 0,2% (p / v) (BSA), 0.1% (v / v) de β-mercaptoetanol (BME) | La mezcla tampón que contiene sólo sacarosa, Tris y EDTA se puede preparar hasta un mes de antelación y se mantiene a 4ºC. PVP, BSA y BME deben añadirse frescos a una alícuota de la cantidad requerida de tampón justo antes de su uso. | Método 1 |
| ST Buffer | Sacarosa 400 mM, Tris 50 mM pH 7,8, polivinilpirrolidona al 0,6% (p / v), albúmina de suero bovino (BSA) al 0,1% (p / v) | La mezcla tampón que contiene sólo sacarosa y Tris se puede preparar hasta un mes de antelación y se mantiene a 4ºC. Obsérvese que el tampón ST no contiene EDTA o BME, y contiene una menor concentración de BSA. | Método 1 |
| Acciones de DNasa | 2 mg / ml de DNasa en NaCl 0,15 M a una concentración de stock de 2 mg / ml | Almacene alícuotas de 200 μl a -20 ° C. Para preparar la solución de trabajo de DNasa (200 μl de solución de DNasa por muestra) véaseTabla 1 a continuación. Vea el protocolo completo abajo para detalles completos de la digestión de DNasa. DNase solución de trabajo debe ser preparado fresco. Para detener la reacción de DNasa se requiere una solución de 400 mM EDTA pH 8,0 (la concentración final necesaria para detener la reacción es EDTA 0,2 M, véase el protocolo completo para más detalles). | Método 1 |
| DNase solución de trabajo | 0,25 mg / ml de DNasa y 20 mM MgCl 2 en ST Buffer | Preparar fresco, 200 ul por muestra. Las concentraciones mostradas son para el volumen de reacción final, por lo que la mezcla: 62,5 l 2 mg / ml de DNasa (basado en volumen de reacción l final de 500), 4 l 1 M MgCl 2 (basado en 200 l volumen de la solución de DNasa), y 133,5 l de tampón de ST para Un volumen final de 200 μl. | Método 1 |
| Lysis Buffer | EDTA 20 mM, pH 8,0; Tris 10 mM pH 7,9; 500 mM Guanidina-HCl; NaCl 200 mM; 1% de Triton X - 100; 0,5 mg / ml de enzimas lisis deTrichoderma harzianum | Mezclar todos los ingredientes excepto para lisar enzimas y almacenar a temperatura ambiente. Las enzimas de lisis deben añadirse frescas a una pequeña alícuota para su uso inmediato. | Método # 2 |
Tabla 1: Recetas de tapones caseros y existencias de trabajo.
| Hoja de Concentración | |||||||
| NOMBRE DE MUESTRA | Peso del dispositivo vacío (g) | Peso del dispositivo llenado (g) | Volumen llenado (ul, lleno menos pesos vacíos) | Peso después de la 1ª vuelta (20 min *, g) | Volumen después de la primera vuelta (ul, llenoMenos los pesos vacíos) | Peso después de la segunda vuelta (X min *, g) | Volumen después de la segunda vuelta (ul, lleno menos pesos vacíos) |
| Tenga en cuenta que el volumen recuperado real será de unos pocos ul menos que el volumen calculado. | |||||||
Tabla 2: Hoja de trabajo de concentración.
| Nombre | Especificidad del genoma | Fuente de Secuencia de Genes | Secuencia (5 '- 3') |
| Ta_ACTIN - F | Nuclear | Andamio de Gramene IWGSC_CSS_1AS_scaff_3272162: 10.663-12.557 | CAGGTATCGCTGACCGTATGA |
| Ta_ACTIN - R | Nuclear | Lo mismo que arriba | GAAGGTAGGGCTGAACAAGAAAC |
| Ta_NAD3 - F | Mitocondrial | Ingreso NCBI EU534409.1 | GGTGATGCCAGAAGTCGTTT |
| Ta_NAD3 - R | Mitocondrial | Lo mismo que arriba | CAGATCAATCTTGTTAGGAGGTACTG |
| Ta_PSBB - F | Cloroplasto | Acceso NCBI KJ592713.1 | GCTACCTTTGCTTTGCTCTTCT |
| Ta_PSBB - R | Cloroplasto | Lo mismo que arriba | GCTGCCTGTTTCCTTGTAGTT |
Tabla 3: Lista de cebadores qPCR.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Hasta la fecha, la mayoría de los estudios de secuenciación organelar se centran en los métodos tradicionales de CC para enriquecer el ADN específico. Métodos para aislar orgánulos de diversas plantas se han descrito, incluyendo el musgo 40 ; Monocotiledóneas tales como trigo 15 y avena 11 ; Y dicotiledóneas tales como arabidopsis 11 , girasol 17 , y colza 14 . La mayoría de los protocolos se centran en el tejido foliar 13 , 14 , 15 , 16 , 17 , algunos de los cuales han sido adaptados para una variedad de tipos de tejidos, incluidas las semillas 11 . El aislamiento de orgánulos de protoplastos también se ha demostrado [ 41] . Sin embargo, esto no es susceptible a todos los sistemas, ni es factible cuando el tejido de interés es limitado. Muchas de estas orgasLos métodos de aislamiento nelar fueron diseñados para recuperar orgánulos intactos para experimentos específicos, tales como estudios fisiológicos. Estos protocolos son engorrosos y típicamente requieren el uso de gradientes de densidad, tales como gradientes de sacarosa o Percoll, que son muy eficientes para aislar fracciones organelares específicas pero que requieren un gran aporte tisular ( es decir, más de 5 gy más de kilogramos, dependiendo de El tipo de tejido). Sin embargo, el método DC se puede optimizar para enriquecer para fracciones celulares específicas, tales como las mitocondrias o el cloroplasto, cambiando velocidades de centrifugación y gradientes de densidad. Por el contrario, el enfoque MF requiere mucho menos material de partida (20 mg), pero los ADN mitocondrial y plastídico estarán presentes por su abundancia relativa en el tejido utilizado para la extracción de ADN. Sin embargo, el protocolo MF ofrece un enfoque alternativo para aislar ADN organelo mixto y es particularmente beneficioso para comenzar con pequeñas cantidades de tejido.
T O evaluar la pureza de la muestra después del aislamiento de organelos, la mayoría de los estudios realizados hasta la fecha sólo utilizan PCR de punto final y electroforesis en gel 11 , 12 . Esto da una medida cualitativa justa de la pureza de la muestra. Sin embargo, los bajos niveles de amplificación pueden no visualizarse en un gel de agarosa. Pocos informes incluyen más medidas cuantitativas de control de calidad, como qPCR 14 . Para una evaluación cuantitativa de la pureza de la muestra de ADN aislada de ambos métodos, se utilizó qPCR y secuenciación para determinar la cantidad de ADN nuclear se mantiene en la muestra, así como las proporciones relativas de ADN mitocondrial versus cloroplasto. Ambos métodos evaluados aquí son eficientes para eliminar el ADN nuclear. Ambos métodos producen una mezcla de ADN mitocondrial y de cloroplasto, aunque en diferentes proporciones.
Cultivar las plantas en la oscuridad (etiolation) se informa para ayudar a facilitar el aislamiento organellar debido a una reducción de fenólicosSin embargo, en esta comparación, no encontramos una ventaja apreciable en el trabajo con tejido etiolado sobre muestras cultivadas de forma ligera. Aunque la proporción de cloroplastos especializados será probablemente mayor cuando se produce luz, el número total de plastidios, Por lo tanto, para los análisis funcionales posteriores, como la evaluación de la heteroplasmia en diferentes tejidos o bajo diferentes factores de estrés o para el análisis de la expresión, se recomienda realizar la secuenciación genómica en el genoma del cloroplasto. Plantas cultivadas bajo condiciones fisiológicamente relevantes.
Para la aplicación con tecnologías de secuenciación de lectura corta, ambas técnicas comparadas aquí producen cantidad y calidad adecuadas de ADN. Sin embargo, para obtener lecturas largas de> 20 kb para aplicaciones de secuenciación de molécula única, es necesaria una mayor cantidad de ADN de mayor calidad. Por ejemplo, idealmente,> 1 μg de orga puraADN de trigo nelar con un peso molecular> 20 kb es necesario para los protocolos internos de baja entrada para preparaciones de biblioteca de insertos de 20 kb 42 . Los nuevos protocolos desarrollados por el usuario y con bajos insumos pueden reducir los requerimientos de ADN ( es decir, 50 ng o incluso menos 20 ), pero el reto sigue siendo tener ADN de alta calidad y alto peso molecular entrando en los preparativos de la biblioteca. Es esencial que la mayor parte del ADN sea> 20 kb, ya que los fragmentos más pequeños se insertarán preferentemente en el SMRTbell y eliminarán la distribución de tamaños de la biblioteca [ 43] . Intentamos una serie de protocolos de extracción de ADN hechos en casa y una serie de protocolos comerciales para la extracción de ADN (no se muestra). Para el tejido de hoja de trigo, el mejor equilibrio entre la cantidad y la calidad del ADN, particularmente la longitud, se obtuvo utilizando un kit comercial 27 , 29 . Dependiendo de las especies de plantas y tejidos de interés, alternatiLos protocolos de extracción pueden ser igualmente adecuados o más fructíferos. No obstante, se concluye que la extracción total de ADN genómico de alto peso molecular de más de 50 kb, seguida de fraccionamiento con la aproximación desplegable MBD2-Fc 28 , es susceptible de secuenciación de larga lectura a partir de material de partida limitado. El trabajo futuro debería probar los límites del material de partida requerido después del fraccionamiento para la preparación de la biblioteca de insertos largos y la secuenciación posterior de lectura larga. Críticamente, este enfoque podría proporcionar un método robusto para aislar el ADN de una submuestra de una única hoja que es adecuado para la secuenciación de larga lectura, sin amplificación del genoma completo. Anticipamos que este enfoque será fácilmente adaptable a otros tipos de tejidos y ampliamente aplicable a otras especies de plantas. Será particularmente útil en situaciones en las que las cantidades de tejido son limitantes, tales como la secuenciación en generaciones individuales en un esquema de cruce o en tipos de tejido más raros.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Los autores declaran que no tienen intereses en competencia.
La mención de nombres comerciales o productos comerciales en esta publicación es únicamente con el propósito de proporcionar información específica y no implica recomendación o aprobación por parte del Departamento de Agricultura de los Estados Unidos. USDA es un proveedor y empleador que ofrece igualdad de oportunidades.
Acknowledgments
Nos gustaría agradecer el financiamiento del Departamento de Agricultura de Estados Unidos - Servicio de Investigación Agrícola y de la National Science Foundation (IOS 1025881 e IOS 1361554). Damos las gracias a R. Caspers por el mantenimiento de invernaderos y el cuidado de las plantas. También damos las gracias al Centro de Genómica de la Universidad de Minnesota, donde se realizaron los preparativos de la biblioteca de Illumina y la secuenciación. También estamos agradecidos por los comentarios de los editores de la revista y cuatro revisores anónimos que fortalecieron aún más nuestro manuscrito. También agradecemos a la OCDE por una beca a SK para integrar estos protocolos para proyectos colaborativos con colegas en Japón.
Materials
| Name | Company | Catalog Number | Comments |
| 2-mercaptoethanol (beta-mercaptoethanol; BME) | Sigma Aldrich | M3148-100ml | |
| 2-propanol (Isopropyl alcohol/isopropanol), bioreagent | Sigma Aldrich | I9516 | |
| agarose, Bio-Rad Cetified Megabase agarose | Bio-Rad | 1613108 | |
| analytical balance | Mettler Toledo | AB54-S | |
| balance | Mettler Toledo | PB1502-S | |
| bovine serum albumin (BSA) | Sigma Aldrich | B4287-25G | |
| Ceramic grinding cylinders, 3/8in x 7/8in | SPEX SamplePrep | 2183 | |
| Cryogenic Blocks compatible with tissue homogenizer for holding 50 mL tubes | SPEX SamplePrep | 2664 | |
| DNaseI | Sigma | DN25 | |
| ethanol, absolute | Decon Laboratories | 2716 | |
| Ethylenediamine Tetraacetic Acid (EDTA), 0.5 M Solution, pH 8.0 | Fisher | BP2482-500 | |
| gel imaging system | |||
| gel stain | Such as GelRed or Ethidium Bromide | ||
| grinding pestle, wide tip for 2 mL conical tubes | |||
| Guanidine-HCl, 8 M solution | ThermoFisher | 24115 | |
| LightCycler 480 SYBR Green I Master | Roche | 4707516001 | |
| liquid nitrogen | |||
| Lysing enzymes from Trichoderma harzianum | Sigma | L1412 | |
| Magnesium Chloride | G Bioscience | 24115 | |
| magnetic rack | ThermoFisher | A13346 | |
| microcentrifuge tubes, LoBind 1.5 mL | Eppendorf | 22431021 | |
| microcentrifuge tubes, standard nuclease-free 1.5 mL | Eppendorf | ||
| microcentrifuge, refrigerated | Sorvall | Legend X1R | Or equivalent product, must be capable of reaching at least 18,000 x g with rotors for 50 mL tubes, Oak Ridge tubes, and 1.5 mL tubes |
| microcentrifuge, room temperature | Eppendorf | 5424 | Or equivalent product, must be capable of reaching at least 18,000 x g with rotor for 1.5 mL and 2 mL microcentrifuge tubes |
| Microcon DNA Fast Flow Centrifugal Filter Units | EMD Millipore | MRCFOR100 | |
| Miracloth, 1 square per sample cut to fit funnel | EMD Millipore | 475855 | |
| NEBNext Microbiome DNA Enrichment Kit | New England Biolabs | E2612L | |
| parafilm | Parafilm M | PM992 | |
| plastic pots and trays | |||
| polyvinylpyrrolidone (PVP) | Fisher | BP431-100 | |
| Proteinase K | Qiagen | 19131 | |
| Pulsed-Field Gel Electrophoresis rig (e.g. CHEF DR III) | Bio-Rad | 1703697 | |
| purification beads, Agencourt AMpureXP beads | Beckman Coulter | A63881 | |
| QIAamp DNA Mini Kit | Qiagen | 51304 | |
| Qiagen 20/g Genomic Tip DNA Extraction Kit | Qiagen | 10223 | |
| Qiagen Buffer EB (elution buffer) | Qiagen | 19086 | |
| Qiagen DNA Extraction Buffer Set | Qiagen | 19060 | |
| QiaRack | Qiagen | 19015 | |
| qPCR machine (e.g. Roche Light Cycler 480) | Roche | ||
| qPCR plate sealing film | Roche | 4729757001 | |
| qPCR plate, 96 well plate | Roche | 4729692001 | |
| Qubit assay tubes | Life Technologies | Q32856 | |
| Qubit Broad Spectrum assay kit | Life Technologies | Q32850 | |
| Qubit High Sensitivity assay kit | Life Technologies | Q32851 | |
| RNaseA | Qiagen | 19101 | |
| Serological pipettes (20 mL) and pipet-aid | Fisher | 13-678-11 | |
| Small funnels, 1 per sample | |||
| Sodium Chloride | Ambion | AM9759 | |
| Soft paintbrush, 2 per sample | |||
| SPEX SamplePrep 2010 Geno/Grinder or another type of tissue homogenizer | SPEX SamplePrep | Or another comparable tissue homogenizer. If you do not have access to a tissue homogenizer, then grinding in a pre-chilled mortar and pestle will suffice (see protocol for details). However, a homogenizer will give more consistent results and total homogenization time is reduced. | |
| Sucrose | Omnipure | 8550 | |
| TBE | |||
| thermomixer | |||
| Tris | Sigma | T2819-100ml | |
| Triton X-100 | Promega | H5142 | |
| tube rotater | |||
| tubes, 50 mL conical polypropylene | Corning | 352070 | |
| tubes, 50 mL high-speed polypropylene | ThermoScientific/Nalgene | 3119-0050 | e.g. Nalgene Oakridge tubes or equivalent |
| vermiculite | |||
| water bath | |||
| water, sterile and certified Nuclease-free | Fisher | 1481 | |
| water, sterile milliQ |
References
- Liberatore, K. L., Dukowic-Schulze, S., Miller, M. E., Chen, C., Kianian, S. F. The role of mitochondria in plant development and stress tolerance. Free Radic Biol Med. 100, 238-256 (2016).
- Samaniego Castruita, J. A., Zepeda Mendoza, M. L., Barnett, R., Wales, N., Gilbert, M. T. Odintifier--A computational method for identifying insertions of organellar origin from modern and ancient high-throughput sequencing data based on haplotype phasing. BMC Bioinformatics. 16 (232), 1-13 (2015).
- Zhang, T., Zhang, X., Hu, S., Yu, J. An efficient procedure for plant organellar genome assembly, based on whole genome data from the 454 GS FLX sequencing platform. Plant Methods. 7 (38), 1-8 (2011).
- Wambugu, P. W., Brozynska, M., Furtado, A., Waters, D. L., Henry, R. J. Relationships of wild and domesticated rices (Oryza AA genome species) based upon whole chloroplast genome sequences. Sci Rep. 5 (13957), 1-9 (2015).
- Iorizzo, M., et al. De novo assembly of the carrot mitochondrial genome using next generation sequencing of whole genomic DNA provides first evidence of DNA transfer into an angiosperm plastid genome. BMC Plant Biol. 12 (61), 1-17 (2012).
- Park, S., et al. Complete sequences of organelle genomes from the medicinal plant Rhazya stricta (Apocynaceae) and contrasting patterns of mitochondrial genome evolution across asterids. BMC Genomics. 15 (405), 1-18 (2014).
- Skippington, E., Barkman, T. J., Rice, D. W., Palmer, J. D. Miniaturized mitogenome of the parasitic plant Viscum scurruloideum is extremely divergent and dynamic and has lost all nad genes. Proc Natl Acad Sci U S A. 112 (27), E3515-E3524 (2015).
- Wicke, S., Schneeweiss, G. M. Chapter 1. Next Generation Sequencing in Plant Systematics. Hörandl, E., Appelhans, M. , Koeltz Scientific Books. (2015).
- Sloan, D. B. One ring to rule them all? Genome sequencing provides new insights into the 'master circle' model of plant mitochondrial DNA structure. New Phytol. 200 (4), 978-985 (2013).
- Woloszynska, M. Heteroplasmy and stoichiometric complexity of plant mitochondrial genomes--though this be madness, yet there's method in't. J Exp Bot. 61 (3), 657-671 (2010).
- Ahmed, Z., Fu, Y. B. An improved method with a wider applicability to isolate plant mitochondria for mtDNA extraction. Plant Methods. 11 (56), 1-11 (2015).
- Ejaz, M., et al. Comparison of small scale methods for the rapid and efficient extraction of mitochondrial DNA from wheat crop suitable for down-stream processes. Genet Mol Res. 13 (4), 10320-10331 (2014).
- Eubel, H., Heazlewood, J. L., Millar, A. H. Isolation and subfractionation of plant mitochondria for proteomic analysis. Methods Mol Biol. 355, 49-62 (2007).
- Hao, W., Fan, S., Hua, W., Wang, H. Effective extraction and assembly methods for simultaneously obtaining plastid and mitochondrial genomes. PLoS One. 9 (9), e108291 (2014).
- Pomeroy, M. K. Studies on the respiratory properties of mitochondria isolated from developing winter wheat seedlings. Plant Physiol. 53 (4), 653-657 (1974).
- Taylor, N. L., Stroher, E., Millar, A. H. Arabidopsis organelle isolation and characterization. Methods Mol Biol. 1062, 551-572 (2014).
- Triboush, S. O., Danilenko, N. G., Davydenko, O. G. A method for isolation of chloroplast DNA and mitochondrial DNA from Sunflower. Plant Mol Biol Rep. 16 (2), 183-189 (1998).
- Pinard, R., et al. Assessment of whole genome amplification-induced bias through high-throughput, massively parallel whole genome sequencing. BMC Genomics. 7 (216), 1-21 (2006).
- Lamble, S., et al. Improved workflows for high throughput library preparation using the transposome-based Nextera system. BMC Biotechnol. 13 (104), 1-10 (2013).
- Raley, C., et al. Preparation of next-generation DNA sequencing libraries from ultra-low amounts of input DNA: Application to single-molecule, real-time (SMRT) sequencing on the Pacific Biosciences RS II. bioRxiv. , (2014).
- Tsai, Y. C., et al. Resolving the Complexity of Human Skin Metagenomes Using Single-Molecule Sequencing. MBio. 7 (1), e01948 (2016).
- Feehery, G. R., et al. A method for selectively enriching microbial DNA from contaminating vertebrate host DNA. PLoS One. 8 (10), e76096 (2013).
- Yigit, E., Hernandez, D. I., Trujillo, J. T., Dimalanta, E., Bailey, C. D. Genome and metagenome sequencing: Using the human methyl-binding domain to partition genomic DNA derived from plant tissues. Appl Plant Sci. 2 (11), 1-6 (2014).
- Noyszewski, A. K., et al. Accelerated evolution of the mitochondrial genome in an alloplasmic line of durum wheat. BMC Genomics. 15 (67), 1-16 (2014).
- Qiagen. QIAamp DNA Mini and Blood Mini Handbook. , 5th ed, Available from: https://www.qiagen.com/ch/resources/ (2016).
- E.M. Corporation. User Guide: Microcon Centrifugal Filter Devices. , Available from: http://www.emdmillipore.com/US/en/product/Microcon-DNA-Fast-Flow-Centrifugal-Filter-Unit-with-Ultracel-membrane,MM_NF-MRCF0R100 (2013).
- Qiagen. User developed protocol: Isolation of genomic DNA from plants and filamentous fungi using the QIAGEN Genomic-tip - (EN). , Available from: https://www.qiagen.com/ch/resources/ (2001).
- New England BioLabs, Inc.. NEBNext Microbiome DNA Enrichment Kit: Instruction Manual Version 4.0. , Available from: http://www.neb.com/~/media/Catalog/All-Products/371BCB5A557C462D95D1E45E15BBFEA3/Datacards or Manuals/E2612Manual.pdf (2015).
- Qiagen. QIAGEN Genomic DNA Handbook. , Available from: https://www.qiagen.com/ch/resources/ (2012).
- PacificBiosciences. Guidelines for Using the BIO-RAD® CHEF Mapper® XA Pulsed Field Electrophoresis System. , Available from: http://www.pacb.com/wp-content/uploads/Unsupported-Guidelines-Using-BIO-RAD-CHEFMapper-XA-Pulsed-Field-Electrophoresis.pdf (2016).
- Andrews, S. FastQC: A quality control tool for high throughput sequence data. , Available from: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (2016).
- Bolger, A. M., Lohse, M., Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 30 (15), 2114-2120 (2014).
- Ogihara, Y., et al. Structural dynamics of cereal mitochondrial genomes as revealed by complete nucleotide sequencing of the wheat mitochondrial genome. Nucleic Acids Res. 33 (19), 6235-6250 (2005).
- Ogihara, Y., et al. Structural features of a wheat plastome as revealed by complete sequencing of chloroplast DNA. Mol Genet Genomics. 266 (5), 740-746 (2002).
- International Wheat Genome Sequencing Consortium (IWGSC). A chromosome-based draft sequence of the hexaploid bread wheat (Triticum aestivum) genome. Science. 345 (6194), (2014).
- Langmead, B., Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat Methods. 9 (4), 357-359 (2012).
- Bendich, A. J. Why do chloroplasts and mitochondria contain so many copies of their genome? Bioessays. 6 (6), 279-282 (1987).
- Kumar, R. A., Oldenburg, D. J., Bendich, A. J. Changes in DNA damage, molecular integrity, and copy number for plastid DNA and mitochondrial DNA during maize development. J Exp Bot. 65 (22), 6425-6439 (2014).
- Ma, J., Li, X. Q. Organellar genome copy number variation and integrity during moderate maturation of roots and leaves of maize seedlings. Curr Genet. 61 (4), 591-600 (2015).
- Lang, E. G., et al. Simultaneous isolation of pure and intact chloroplasts and mitochondria from moss as the basis for sub-cellular proteomics. Plant Cell Rep. 30 (2), 205-215 (2011).
- Tobin, A. K. Subcellular fractionation of plant tissues. Isolation of chloroplasts and mitochondria from leaves. Methods Mol Biol. 59, 57-68 (1996).
- PacificBiosciences. Procedure & Checklist - 10 kb to 20 kb Template Preparation and Sequencing with Low (100 ng) Input DNA. , Available from: http://www.pacb.com/wp-content/uploads/Procedure-Checklist-10-20kb-Template-Preparation-and-Sequencing-with-Low-Input-DNA.pdf (2015).
- PacificBiosciences. Template Preparation and Sequencing Guide. , Available from: http://www.pacb.com/wp-content/uploads/2015/09/Guide-Pacific-Biosciences-Template-Preparation-and-Sequencing.pdf (2014).
