

Summary
Utnyttja en förmonterade Cas9 är komplexa ribonukleoprotein (RNP) en kraftfull metod för precisa, effektiva gen editering. Här, belysa vi dess användbarhet inom ett brett spektrum av celler och organismer, inklusive primära mänskliga celler och både klassiska och nya modellorganismer.
Abstract
Platsspecifika eukaryota gen editering med CRISPR (klustrad regelbundet mellanliggande kort palindromic repetitioner)-Cas (CRISPR-associerade) system har snabbt blivit ett vanligt bland forskare bedriver en mängd olika biologiska frågor. Användare använder oftast den Cas9 protein från Streptococcus pyogenes i ett komplex med en lätt omprogrammeras guide RNA (gärna). Dessa komponenter introduceras in i celler, och i en base ihopkoppling med en kompletterande region av dubbelsträngat DNA (dsDNA) genomet, enzymet klyver båda delarna för att generera en dubbel-strand paus (DSB). Efterföljande reparation leder till antingen slumpmässig införande eller borttagning händelser (indels) eller införlivandet av förutsatt att försöksledaren DNA på platsen för avbrottet.
Användning av ett renat singel-guide RNA och Cas9 protein, förmonterade för att bilda en RNP och levereras direkt till celler, är en potent strategi för att uppnå högeffektiva gen redigering. RNP redigering ökar särskilt graden av genen införande, ett resultat som ofta är svårt att uppnå. Jämfört med leverans via en plasmid, leder kortare varaktigheten av den Cas9 RNP i cellen till färre off-target händelser.
Trots sina fördelar är många casual användare av CRISPR gen redigering mindre bekant med denna teknik. För att sänka inträdeshindret, beskriver vi detaljerade protokoll för genomförandet av RNP-strategi i en rad sammanhang, belysa dess distinkta fördelar och applikationsmöjligheter. Vi täcker redigering i två typer av primära mänskliga celler, T-celler och hematopoetiska stamceller/stamceller (HSPCs). Vi visar också hur Cas9 RNP redigering gör lättköpt genmanipulation av hela organismer, inklusive klassisk modell rundmasken Caenorhabditis elegans och mer nyligen infört modellen kräftdjur, Parhyale hawaiensis.
Introduction
Hotellets CRISPR-Cas9 systemet tillåter forskare att förändra riktade regioner av någon genomet1. Denna snabb och billig teknik har revolutionerat grundforskning och lovar att göra en djupgående inverkan på utvecklingen av personlig sjukdom terapier, precisionsjordbruk, och bortom2. CRISPR redigering är en demokratiserande verktyg och genomföra systemet i ett nytt laboratorium kräver ingen särskild expertis i genomet engineering, bara grundläggande molekylärbiologi färdigheter. Forskare kan nu studera tidigare svårlösta organismer med några alternativa sätt för genmanipulation3,4. Enbart under de senaste fem åren, har CRISPR gen editering använts till ingenjör över 200 olika ryggradsdjur, ryggradslösa djur, växt och mikrobiell arter.
Anpassad från CRISPR prokaryota försvar vägen, de centrala delarna krävs för platsspecifika gen editering är proteinet Cas9, vanligtvis från S. pyogenes och kodon-optimerade med ljussignal added nukleära lokalisering (NLS) och dess specialiserade RNA guide5,6. Även om inte diskuteras här, andra Cas9 orthologues eller CRISPR endonucleases kan också användas. Den naturligt förekommande gärna består av två separat transkriberas bitar, CRISPR RNA (crRNA) och den trans-aktivera crRNA (tracrRNA)7. Dessa RNAs kan vara smält till en enda utskrift, känd som den enda-guide RNA (sgRNA)8. De flesta genomet redaktörer välja den strömlinjeformade sgRNA9, även om den dubbla-guiden också används regelbundet10,11. Praktiker välja 20-nukleotid (nt) genomisk DNA mål, se till att det ligger intill en kort licensiering signatur krävs för Cas9 erkännande, kallas en protospacer intill motiv (PAM), och utforma en gärna som innehåller sekvensen kompletterande12 .
En gång inne i cellen, den komplexa RNP lokaliserar dess genomisk mål, gärna basparen med kompletterande DNA strand och sedan enzymet klyver bryta båda DNA-strängar att generera en dubbel-strand2. Cell reparation maskiner fixar Tvistlösningsorganet av en av minst två vägar: via felbenägen icke-homolog slutet-anslutning (NHEJ) vägen eller homologi-regisserad reparation (HDR), som sömlöst innehåller DNA som innehåller 'vapen' av homologi endera sidan av avbrottet. Den tidigare reparation vägen leder vanligtvis till ditsatta bildandet och åtföljande gen störningar, medan den senare ger praktiker infoga eller ändra DNA sekvenser1.
Redigering effektiviteten och precisionen är beroende av de medel som Cas9 och gärna in i cellen. Dessa komponenter kan levereras till odlade celler, embryon eller organismer i form av nukleinsyror eller som en förmonterade RNP komplexa13,14,15. Gemensamma nukleinsyrebaserade leveransmetoder inkluderar den virala transduktion, transfection eller elektroporation mRNA eller plasmid DNA. Cas9 protein och guide RNA produceras sedan i cellen och de förknippar bildar ett komplex.
Direkt leverans av RNP kräver separat rening av Cas9 protein och guide RNA. Detta kan göras internt eller protein och sgRNA kan köpas från en av flera kommersiella leverantörer. När förvärvade den Cas9 och gärna blandas för att bilda komplexet RNP enzymatiskt-kompetenta och infördes till celler genom direkt injektion i befruktade ägg och embryon, lipid-baserade transfection16eller elektroporation. Den första rapporten av RNP redigering inblandade injektion i C. elegans gonader17. Mikroskop är medel som föredras för att införa RNP i embryon och hela organismer, om effektiv elektroporation har visats i18,19 och råtta20 musembryon. Vi beskriva protokoll för direkt injicera RNP i C. elegans gonader och P. hawaiensis embryon och rekommenderar en specialiserad typ av elektroporation att leverera RNP när du redigerar primära mänskliga celler. Denna metod, nucleofection, innebär optimerad elektroporation program och cell typspecifika lösningar och tillåter RNP ange både cytoplasman och nucleus21.
Gen editering med RNP erbjuder flera distinkta fördelar. Eftersom protein och RNA komponenter är förmonterade och kvaliteten kan säkerställas före leverans, undviker RNP redigering många fallgropar kopplade nukleinsyra-baserade. Nämligen, det finns ingen risk för Cas9-encoding DNA integrering i den mottagande arvsmassan, mRNA utsätts aldrig för nedbrytning och det kringgår problem med i vivo gärna eller protein, vikning, och föreningsfrihet22,23. Dessutom leder använder RNP till lägre toxicitet och långt färre off-target händelser än plasmid-baserade uttrycket som ett resultat av den RNP kortare halveringstid inuti cellen24,25,26,27.
Slutligen, RNP redigering bevisligen leder till hög redigering priser i en mängd olika mänskliga cellinjer, primära celler som fibroblaster, embryonala stamceller (EKSG), inducerade pluripotenta stamceller (iSPCs), HSPCs, och T cells16,24, 25,26,27,28,29; hos ryggradslösa djur inklusive C. elegans, P. hawaiensisoch bananflugor3,17,30; i ryggradsdjur som zebrafisk, möss och råttor31,32. i växtart inklusive Arabidopsis, tobak, sallad, ris, grapevine, apple, majs och vete33,34,35,36. och i Chlamydomonas, Penicilliumoch Candida arter37,38,39. Frekvensen av ditsatta bildandet kan vara högre vid användning av RNP jämfört med plasmid leverans och HDR-medierad DNA införande kan bli lättare att uppnå25,27,29.
Protokollet beskrivs här använder den Cas9 RNP och är en effektiv, lätt anpassningsbar teknik som är enkel att tillämpa på en mängd olika biologiska system40,41, särskilt i celler som annars är svårt att arbeta med och i organismer utan väletablerade system för exakt genetisk manipulation. Vi börjar med att beskriva hur man designar, erhålla och montera den Cas9 RNP innan täcker dess användning över olika modell celltyper och organismer. Hematopoetiska stamceller/stamceller celler (HSPCs) och T-celler redigeras med samma metod, nucleofection, så de täcks tillsammans i steg 2 och 3 i detta protokoll. Redigera förfaranden för C. elegans beskrivs i steg 4 och 5, och P. hawaiensis redigering är täckt i steg 6 och 7. Slutligen, eftersom framgången för ett genredigering experiment i någon organism kan bedömas av genotyp sekvensering, delsteg som beskriver möjliga analysmetoder för alla celler och organismer beskrivs i protokollet anges i steg 8.
Protocol
1. RNP församling
-
Utforma experimentet i god tid, förvärva alla RNA, DNA och protein komponenter före tid. Som ett första pass, prova en av de positiva kontrollerna listade i tabell 1 och kommersiella reagenser beskrivs i tabell av material används för att säkerställa en tillförlitlig experimentell design och integriteten av material. För ytterligare tips om hur du planerar ett nytt editering experiment, se papper på detta ämne12,42,43.
Obs: När monterad som beskrivs i de efterföljande stegen, RNPs förberett i förväg kan förvaras vid-80 ° C.- När du har valt vilken gen att rikta, använda en av de gratis online verktyg för att utforma en optimal gärna44,45,46,47,48. Var noga med att rikta ett exon om hoppas att generera en knockout.
Obs: Dessa verktyg hjälper till att identifiera en målwebbplats med en intilliggande S. pyogenes PAM sekvens, hög kvalitet poäng och låg off-target poäng. - Rena S. pyogenes Cas9 proteinet genom publicerade metoder8, eller köpa den från en kommersiell leverantör.
- Förbereda en typisk Cas9-buffert för RNA utspädning, RNP förberedelse och protein lagring, som innehåller 20 mM HEPES pH 7.5, 150 mM KCl, 10% glycerol och 1 mM av TCEP. Använd alltid nuclease-fritt vatten i buffertar som används för att resuspendera eller späd RNA för att förhindra nedbrytning.
- Producera guiden RNA (tracrRNA och crRNA eller sgRNA) genom en in vitro- transkription med publicerade metoder, eller köpa den från en nukleinsyra syntes företaget17,21,49, 50 , 51.
- Om du sätter en gen, syntetisera eller köpa en givare DNA mall.
- Lagra protein och RNA alikvoter vid-80 ° C och Tina på is omedelbart före användning.
Obs: Varje frysning-tining något sänker effektivitet. Detaljerad, öppna protokoll för Cas9 rening52 och in vitro- transkription av sgRNAs53 finns tillgängliga någon annanstans.
- När du har valt vilken gen att rikta, använda en av de gratis online verktyg för att utforma en optimal gärna44,45,46,47,48. Var noga med att rikta ett exon om hoppas att generera en knockout.
- Om arbetar med C. elegans, hoppa till steg 1,5. För protokollet P. hawaiensis , hoppa till steg 1,6. Om du använder sgRNA, hoppa till steg 1.4. Fortsätt till steg 1.3 för att montera en gärna för primär cellredigering.
-
Montera en gärna genom att blanda equimolar mängder av tracrRNA och crRNA. Gör 100 µL av 80 µM gärna lager, för ca 50 genomet redigering experiment.
- Inkubera gärna vid 37 ° C i 30 min och låt det långsamt svalna till rumstemperatur.
-
RNP prep för HSPC och T cellredigering: montera en RNP komplexa genom att blanda en 1-2 x molar mängd gärna till 200 pmol Cas9 protein i en total volym av 10 µL. mycket långsamt, Lägg koncentrerad Cas9 till den gärna (pre utspätt i Cas9 bufferten) för ca 30 s , att göra snabba cirklar med pipetten, föra den slutliga Cas9-koncentrationen till 20 µM.
- Förbereda de elektroporation kyvetter.
Obs: Detta protokoll är specifika för det kommersiella systemet som avses i Tabell av material, men RNP redigering kan också uppnås med andra elektroporation enheter. - Tillsätt 5 µL (100 pmols, T-celler) eller 10 µL (200 pmol, HSPCs) RNP till varje kyvetten.
- Om du sätter nytt DNA i stället för att göra en knockout, lägga 1 µL 100 µM (100 pmol) enkelsträngat oligonukleotiden givare DNA (ssODN)25,54,55 till kyvetter eller brunnar i plattan.
- Hoppa till steg 2 för nästa instruktionerna i primär cellredigering protokollet.
- Förbereda de elektroporation kyvetter.
-
RNP prep för C. elegans redigering: montera RNP komplexa genom att lägga till följande reagenser för att skapa en slutlig volym av 20 µL (slutliga halterna noteras inom parentes): Cas9 (2 µM), HEPES pH 7.5 (10 µM), KCl (115 µM), crRNA (12 µM) , tracrRNA (40 µM), och den reparation mallar om det behövs (0,5 µM ssDNA eller upp till 350 ng/µL dsDNA).
Obs: Effektiviteten av en Cas9-medierad DSB-mallade reparation är proportionell mot koncentrationen av den dsDNA reparation bygga; alltså, ju högre koncentration av mallen reparation, ju effektivare mallade reparation. En injektion av blandningar som innehåller mer än 350 ng/µL av dsDNA har dock visat sig minska lönsamheten för injicerade maskar. Således är det bäst att använda upp till, men inte mer än 350 ng/µL av dsDNA i mixen för att maximera reparation effektivitet och minimera dess dödlighet.- Lägga till flera crRNAs att rikta flera loci samtidigt, som behövs för den co-CRISPR/co-conversion screening metod som beskrivs i steg 5,4. När du lägger till fler än en crRNA, lägga till varje sekventiellt till huvudmixen.
Obs: Mängden varje crRNA behöver inte vara samma, och även fördubbla den totala koncentrationen av crRNAs i huvudmixen utan att ändra koncentrationen av Cas9 verkar inte störa frekvensen av mutagenes på en specifik locus. Exempel är beskrivs i detalj i Paix et al. 56. - Blanda genom pipettering och snurra RNP lösningen vid 16 000 x g i 5 s för att säkerställa att lösningen samlas på botten av röret.
- Inkubera lösningen vid 37 ° C under 15 m.
- Centrifugera provet vid 16 000 x g för 1 min till pellet eventuella partiklar som kan täppa till tunna-uttråkad Mikroskop nålen. Använda supernatanten i efterföljande steg.
- Hoppa till steg 4 för återstoden av protokollet C. elegans .
- Lägga till flera crRNAs att rikta flera loci samtidigt, som behövs för den co-CRISPR/co-conversion screening metod som beskrivs i steg 5,4. När du lägger till fler än en crRNA, lägga till varje sekventiellt till huvudmixen.
-
RNP prep för P. hawaiensis redigering: förbereda engångsbruk Cas9 alikvoter genom att späda dem med nuclease-gratis vatten och fenolrött (för att visualisera injektioner) till en slutkoncentration av 6,25 µM av Cas9 och 0,15% fenolrött.
- Montera RNP komplexa genom att blanda en 2-5 x molar överskott av gärna till Cas9 proteinet i en total volym på 6 µl. Lägg 12 pmol av Cas9 till gärna, att föra slutliga Cas9 koncentration till 2 µM, gärna koncentration till 4-8 µM och fenolrött koncentration på 0,05%.
- Inkubera blandningen i rumstemperatur i 10 min till komplexa RNP.
- Hoppa till steg 6 för nästa instruktionerna i P. hawaiensis redigeringprotokollet.
2. cell kultur och förberedelse
Obs: Utför steg 2.1.1 till 3.3.3 i biologiska säkerhetsdragskåp.
-
Köp frysförvarade mänskliga mobiliserade perifert blod CD34+ HSPCs från en leverantör.
- Tina ~ 1 x106 HSPCs i en 37 ° C vatten bad i 3 min och överföra dem till en 15 mL koniska rör. Tillsätt 10 mL av ett serumfritt expansion medium från en kommersiell källa och snurra blandningen vid 100 x g för 10 min. avlägsna supernatanten och återsuspendera cellerna i 2 mL kompletterade SFEM. Platta celler i 6-bra plattor och kultur dem i en 37 ° C inkubator för 24-48 h innan den RNP elektroporation.
- Räkna cellerna med en hemocytometer och överföra det totala antalet HSPCs behövs (150.000-200.000 HSPCs per kyvetten vara electroporated) till ett centrifugrör.
- Snurra röret vid 100 x g i 10 min till pellet cellerna.
-
Köpa mänskliga primär CD4+ T celler från en leverantör eller isolera dem från humant helblod av täthet lutning centrifugering29.
- Innan T-cellsaktivering, pre coat 48-väl kultur plattor med αCD3 (UCHT1) och αCD28 (CD28.2). Täck plattorna med 500 µL 10 µg/mL αCD3 och 10 µg/mL αCD28 i PBS för minst 2 h vid 37 ° C.
Obs: För vissa lokus, NHEJ kan uppnås utan före stimulering, men inklusive detta steg maximerar dess effektivitet. - Kultur T celler för 48 h vid 37 ° C på αCD3/αCD28 antikropp-bundna tallrikar i RPMI komplett medium [RPMI-1640 kompletteras med 5 mM HEPES, 2 mM av kommersiella alternativ till L-glutamin, 50 µg/mL penicillin/streptomycin, 50 µM av 2-merkaptoetanol, 5 mM icke-essentiella aminosyror, 5 mM av natrium pyruvat och 10% (vol/vol) FBS]. Kultur T celler med en täthet av 2.000.000 T-celler i 500 µL media per brunn 48-bra platta.
- Räkna T celler med hjälp av en hemocytometer och överföra experimentera det totala antalet T-celler som är nödvändiga för elektroporation (100.000-1,000,000 T celler per kyvetten vara electroporated) till ett centrifugrör.
- Snurra röret vid 90 x g i 8 min till pellet cellerna. Om cellerna har täthet lutning-separerad inom 2 dagar, snurra dem på 200 x g i 8 min.
- Innan T-cellsaktivering, pre coat 48-väl kultur plattor med αCD3 (UCHT1) och αCD28 (CD28.2). Täck plattorna med 500 µL 10 µg/mL αCD3 och 10 µg/mL αCD28 i PBS för minst 2 h vid 37 ° C.
-
För båda celltyper, Aspirera supernatanten med pipett/vakuum, ta bort eventuella bubblor.
- Försiktigt resuspendera cellerna med 20 µL av elektroporation buffert per kyvetten.
- Tillsätt 20 µL av cellerna (150.000-200.000 HSPCs eller 100.000-1,000,000 T-celler) i varje kyvetten, som redan innehåller 10 µL av RNP, och blanda väl genom pipettering upp och ner utan att skapa bubblor.
3. RNP elektroporation
- Electroporate kyvetter efter att placera dem i en nucleofector. För HSPCs, använda puls koden ER100. För T-cellerna, använda puls kod EH-115.
-
HSPCs bara: Tillsätt 100 µL av en kompletterade SFEM medium (värmt till 37 ° C) till varje kyvetten omedelbart efter elektroporation och låta cellerna återhämta för 10-15 min.
- Överföra cellerna till kultur dem i en 96-väl runda-botten plattan och lägga till en ytterligare 100 µL av det kompletterade SFEM mediet för 24 h.
- Ändra dem till ett färskt kompletterade SFEM medium och inkubera dem för en ytterligare 24-72 h.
- Ta bort cellerna för genotypning dem 48-96 h post-elektroporation. Snurra cellerna vid 300 x g i 5 min och avlägsna supernatanten innan du börjar den DNA-extraktionen (steg 8,2).
-
T celler endast: lägga till 80 µL av RPMI slutföra kultur media före värmas till 37 ° C från reservoaren till varje kyvetten eller brunn, med flerkanalig pipett (om nödvändigt).
- Inkubera dem vid 37 ° C i 15 min.
- Lägg till lämpliga medier, antikroppar, cytokiner, etc. till den destination skylt(ar) och pre värma dem i en 37 ° C inkubator.
- Överföra 107 µL av electroporated celler från brunnarna till en runda-96 brunnar bottenplatta med flerkanalig pipett (om nödvändigt).
- För information om bedömningen av redigering utfallen, hoppa till steg 8.
4. C. elegans förberedelse
-
1 dag före Mikroskop: förbereda agaros kuddar för Mikroskop.
- Gör en 3% (w/v) agaros lösning i vatten genom att lägga agaros vattnet och föra lösningen till en koka på en värmeplatta eller i mikrovågsugn.
- Ordna 24 x 50 mm x 1,5 mm skyddsglaset diabilder på ett bord och Använd ett glas Pasteur-pipett för att placera en liten (~ 15 µL) droppe agaros lösning in i bilden. Snabbt platta agaros rullgardinsmenyn genom att placera en annan täckglas på toppen. Tillåta Agarens stelna och sedan ta bort en av coverslips.
- Lämna en bordsskiva agaros-belagd täckglas ansikte-upp över natten för att torka. Efter 24 h, lagra agaros dynorna i en ren och torr behållare.
Obs: Dessa kan användas på obestämd tid.
- Dra de Mikroskop nålarna: använda borosilikatglas kapillärer med filament (yttre diameter 1,0 mm och inre diameter 0,58 mm), dra nålar baserat på Mello och brand57 och andra resurser58. Nålarna kan användas omedelbart eller kan lagras i en ren, torr behållare, stagade av lera stöder.
- För underhåll av maskar, förbereda en nematod Growth Media (NGM) agar hälls i Petri plattor och fläckig med OP50 bakterier (för protokoll på standard C. elegans underhåll och recept för tillväxt medier, se Stiernagle59).
- Stage maskar för Mikroskop: 12-24 h innan Mikroskop, plocka L4-iscensatt hermafroditer till en ny NG-agarplatta med OP50 bakterier och inkubera dem över natten vid 20 ° C. För varje Cas9 mål/insprutning mix, plocka ~ 30 maskar till plattan.
-
Dagen i Mikroskop: Ladda drog Mikroskop nålen med den RNP-lösning som beretts i steg 1,5 supernatant.
- Pipettera supernatanten från 1.5.4 steg till en drog kapillär pipett och återfyllning lösningen från kapillär pipetten i beredda Mikroskop nålen (lastning generellt mindre än 0.1 µL).
- Montera den inlästa injektionsnålen på Mikroskop apparaten kopplad till en micromanipulator. Ange apparater Injekteringstryck till 250 kPa och balans trycket till 25 kPa.
-
Bryta tillbaka inlästa nålspetsen att generera en vass nål kant. Placera en 15 x 15 mm x 1,5 mm fyrkantig täckglas på toppen av en 24 mm x 50 mm x 1,5 mm täckglas.
- Överlagra en kant på det fyrkantiga täckglaset med halocarbon olja 700.
- Placera nålen i oljan, vid kanten av de 15 mm fyrkantig täckglaset.
- Använda en hand för att vägleda den Mikroskop scenen och täckglas, borsta bilden upp och längs kanten av nålen medan deprimerande pedal/injektionsknappen. Bryta nålspetsen tillbaka, öka flödet av vätska ur nålen. Uppnå en optimal flödeshastighet genom injektionen blanda flöde längs kanten av nålen, bildar ~ 1 bubbla/s.
- Bekräfta att L4 maskar plockade 12-24 h före Mikroskop är utvecklingsmässigt stegvis unga vuxna på dagen för injektion. Plocka de unga vuxna maskarna till en NG-agarplatta som saknar OP50 bakterier och tillåter dem att krypa runt i 5 min. Detta minskar mängden bakterier överförs till injektion pad, minimera kanylen täpps.
- Placera en agaros injektion pad/täckglas på en dissektion omfattning. Använda en mask plocka, låg en liten koll på halocarbon olja längs ena kanten av kudden.
-
Använder den mask plocka belagd i olja, lyft flera maskar av NG-agar plattan och i spåret av olja. Med ett fint hår bifogas en pipett, såsom en ögonfrans eller katt morrhår, position maskar i parallell, försiktigt trycka maskar i agaros pad. Tills bekväma med förfarandet för Mikroskop, bara montera och injicera en mask i taget.
Obs: Dry Agarens kommer att transportera fukt från maskar, orsakar dem att hålla sig till pad. Följaktligen måste man arbeta snabbt som maskar kan ut.- En gång i placera och fäst till pad, overlay maskar med en annan några droppar halocarbon olja (~ 20 µL) från spetsen av masken plocka.
5. C. elegans Gonad Mikroskop med RNPs och efter injektion hand
Obs: Mikroskop protokollet är anpassad från Mello och brand57och beskrivs i detalj på annan plats60,61.
-
Placera täckglaset med monterade maskar på mikroskopet injektion. I en låg förstoring (5 X mål, 10 X okulär), placera maskar vinkelrätt mot injektionsnålen.
- Växla till en hög förstoring (40 X-objektiv, 10 X okulär), flytta nålen intill gonad armen motsvarar regionen nära atomkärnor i mitten av - till sent-pachytene.
- Använda micromanipulator, flytta nålen mot masken, deprimerande nagelbanden något. Tryck sedan på sidan av Mikroskop scenen för att skaka nålen genom nagelbanden med en hand. Tryck ner pedalen/injektionsknappen och sakta fylla gonad armen med injektion blanda och ta bort nålen.
- Upprepa detta med andra gonad armen.
-
När maskarna injiceras, ta bort täckglas/agaros pad och placera den i dissekera Mikroskop.
- Med dragna kapillär pipett avlägsna oljan från maskar genom att en M9 buffert över dem med. Utföra denna behandling för att släppa maskar från ägarn.
- Efter 10 min, när maskar stryk runt i bufferten, flytta dem till en NG-agarplatta med OP50 bakterier använder drog kapillär pipetten. Placera plattan vid 20 ° C för 2-3 h tills maskar har återhämtat sig och flyttar runt.
- När återvunna, individuellt överföra maskar till NG-agar plattor med OP50 och föra plattorna till en 25 ° C inkubator.
-
Tillåta P0-injicerade maskar växa och lägga avkomma i 3 dagar. Skärmen F1 avkomma.
- Om du använder co-CRISPR eller samtidig konvertering62,Välj63,64,65, kandidat maskar för screening baserat på om de har den mutanta fenotypen av Referensgenen. Individuellt överföra dessa markerade maskar till nya NG-agar plattor med OP50 och tillåta dem att lägga F2 avkomma vid 20 ° C.
Obs: Fenotypen används för en co-CRISPR screening eller urval bör ge en tidig uppskattning för framgång Cas9 redigering. - Om den co-CRISPR fenotypen inte finns, microinject en positiv kontrollplasmid att hjälpa effektivisera Mikroskop.
Obs: till exempel, inklusive en plasmid i injektion mix som kodar mCherry-taggade MYO-2 hjälper bedöma injektion effektiviteten. Maskar som framgångsrikt injiceras med pCFJ90 kommer att ha några avkomma med fluorescerande pharynxes.
- Om du använder co-CRISPR eller samtidig konvertering62,Välj63,64,65, kandidat maskar för screening baserat på om de har den mutanta fenotypen av Referensgenen. Individuellt överföra dessa markerade maskar till nya NG-agar plattor med OP50 och tillåta dem att lägga F2 avkomma vid 20 ° C.
- Undersöka F1 maskar för förekomst av önskade redigeringar. Plocka F1 mor till en enskild brunn en plattan med 96 brunnar, lyse henne och undersöka hennes DNA genom antingen infoga-specifika PCR-amplifiering, DNA-sekvensanalys eller surveyor nuclease assay (CEL-1)66.
Obs: Dessa analyser kan utföras med en co-CRISPR/co-conversion eller andra screening eller urval regimer65,66,67,68. - För information om bedömningen av redigering utfallen, hoppa till steg 8.
6. P. hawaiensis beredning
- 1 dag före Mikroskop, berika för de tidiga embryona genom att inrätta en 'par tank' kvällen innan; Nyligen separerade kvinnor kommer att innehålla färska-befruktade embryon. Se Rehm o.a. 69 för detaljer.
- På dagen för Mikroskop, samla de encelliga Parhyale embryona (0-4 h efter befruktning) av anesthetizing dräktiga honor med 0,02% kryddnejlika olja i havsvatten och försiktigt skrapa embryon ur hennes ventrala barnaskara påse med en flamma-draget och rundad glas pipett och en tråkig par #3 pincett.
7. P. hawaiensis Embryo Mikroskop med RNPs och efter injektion hand
- Återfyllning ovan ett drog kapillärrör med ca 1 µL av RNP injektion mixen.
-
Använd komprimerade kväve till microinject varje embryo som beskrivs i Rehm o.a. 69.
- Injicera Parhyale embryon i dissekera Mikroskop med hjälp av en microinjector och en micromanipulator. Ladda 1.5 µL av injektion mixen på baksidan av en drog kapillärrör (4 inches - 1,0 mm med filament, drog med hjälp av en mikropipett dra apparater) använder en microloader pipettspetsen.
- Ställ in nålen på injektion apparaten och bryta den spetsen av nålen (en mycket liten mängd) använder ett par pincett dissekera omfattas. Kalibrera den volym som levereras genom att injicera i halocarbon olja 700 och mäta diametern på bubblan.
- Skär ett 'tråg' ur den härdare med ett rakblad. Fyll den halvvägs med filter-steriliseras havsvatten, och linje Parhyale embryon i tråg att stabilisera.
- Injicera embryon med Mikroskop setup, stabilisera varje embryo med ett par pincett under injektionen. Efter injektionen, Använd glas överföring pipett för att överföra embryon till en fräsch 60 mm kultur maträtt fylld till hälften med filter-steriliseras havsvatten.
-
Om första divisionen har redan inträffat att bilda ett 2-cell embryo (4-6 h efter fertilisering), generera fullt-mutant djur genom att injicera både blastomerer. För att säkerställa en total klyvning av i 2-cellstadie, Co injicera blastomerer med FITC eller TRITC dextran och Observera att signalen är begränsad till en enda blastomerer under en fluorescerande dissekera omfattning efter injektionen.
- Alternativt kan generera 'halv-mutant' djur genom att injicera bara en av de två blastomerer i 2-cellstadie (grovt delade vänster-höger beroende på vävnad och position längs A-P-axeln).
- Injicera en cell i ett 8-cell embryo (7,5-9 h efter fertilisering) att begränsa redigering till en enda groddblad. Se Gerberding o.a. 70 för en karta över tidiga blastomerer härstamningar.
-
Inkubera embryon i 60 mm kultur rätter (inte mer än 25 per maträtt), fylld till hälften med filter-steriliseras havsvatten, 'syresatt pre' med ett akvarium bubbelflaskan eller genom kraftig skakning.
- Placera rätterna av embryon i ett löst förslutna plasticware fodrad med våta pappershanddukar att behålla fuktighet och placera dem i en 26 ° C inkubator med en 12 h ljus-mörker cykel.
- Överföra de överlevande embryona till rent havsvatten rätter varje några dagar.
Obs: Embryon kan vara odlade i rumstemperatur, även om de kommer att utvecklas mycket långsammare.
-
Dissekera och fixa embryon i olika skeden för uttryck analys av i situ hybridisering eller antikropp färgning (se Browne et al. 71 för en mellanlagringsplats guide och ytterligare referenser för dissektion och fixering72, i situ -hybridisering-73och antikropp färgning74).
- Gör dissektion nålar av gängning en böjd träbit volframtråd cirka 0,5 i längd i änden av en insulin nål. Slipa nålen i natriumhydroxid under en ström. Använd en 1 mL spruta som handtaget på dissektion nålen.
- Fyll en väl av en 3-väl glasskål halvvägs med en nygjord lösning av 9 delar PEM buffert (0.1 M rör pH 6,95, 2 mM av EGTA, 1 mM MgSO4), 1-del 10 x PBS och 1 del 32% PFA. Placera 3-5 embryon i skålen och peta ett litet hål i varje embryo, med en skarp Volfram nål till säcken och med en något slöa för att stabilisera, låta äggulan flöda ut och fixeringsvätskan att köra i.
- Använder ett par slipade volfram nålar, försiktigt reta bort de yttersta två membran som omger Parhyale embryot. Dissekera dem i fixativ för att göra embryon mer robust men arbetar snabbt för att hålla membranet från att bli fast för embryot, vilket försvårar membran borttagning. Tillåta att embryon som fix för totalt 15-20 min. för antikropp färgning eller 40-50 min för i situ hybridisering.
- Bild levande ungar och analysera dem för morfologiska och beteendemässiga fenotyper eller fixa och färga dem för mer detaljerade analyser. Höja ungarna till könsmognad i 2-3 månader att upprätta knockout och transgena linjerna (se Kontarakis och Pavlopoulos75 för hatchling vård och andra användbara Detaljer).
8. bedömning av redigering utfall
- I tillämpliga fall, leta efter en visuell eller funktionella fenotyp i redigerade celler eller organismer.
Obs: Den här processen varierar kraftigt genom ansökan, och några exempel beskrivs i slutet av deras relevanta protokollstegen ovan. Efter korrigering av sickle cell mutation i HSPCs, analysera hemoglobin produktion genom differentierade erytroblaster med HPLC (figur 1A). En knockout av IL-2 receptorn gen i T-celler kan bekräftas genom ytan färgning och flödescytometri (figur 1B). För att bedöma C. elegans och P. hawaiensis fenotyper, iaktta de djurs morfologi och beteende under en ljus eller fluorescerande Mikroskop (siffrorna 1 c och 1 D). - För att avgöra effektiviteten och typ av genomisk redigeringar genereras, lyse pooler av redigerade celler och extrahera deras genomiskt DNA med hjälp av en kommersiell utvinning kit21.
-
För en snabb uppskattning av ditsatta bildandet, PCR-Förstärk minst 200 baspar runt snittet plats och utföra en T7-endonuclease1 (T7E1)76 eller besiktningsman (CEL-1 nuclease) assay77.
- Om en ditsatta formation på Cas9-cut plats eller framgångsrika HDR kommer att skapa eller ta bort en känd begränsning webbplats, överväga att använda en restriktionsenzym matsmältningen för att uppskatta den redigering effektivitet6. Begränsning fragment längd polymorfism (RFLP) analysen kan vara ett praktiskt sätt att kontrollera hur effektiva om det råkar finnas.
- För en exakt kvantifiering av redigering effektivitet och bestämning av dominerande redigering utfall, skicka den PCR-Amplikonen för en standard Sanger sekvensering med både framåt och bakåt grundfärger.
Obs: Om analysera en enda klon eller organismen, analys av Sanger resultat är enkel, som visas i figur 2A. Om analysera en pool av celler, sedan analysera kromatogram med onlineverktyg78, som visas i figur 2B. - För en fullständig kvantifiering och sekvenser av redigering utfall, utföra djupsekvensering27,54, som avbildas i figur 2C.
- Att bedöma en viss uppsättning off-target förändringar, PCR-Förstärk de förutspådda off-target platserna och skicka dem för NGS. För att möjliggöra detektion av kromosomala flyttningar, utföra GUIDE-seq79 eller hög genomströmning, genome-wide translokation sekvensering (HTGTS)80. För en fullständig bild av off-target redigeringar i en klonal befolkning, utföra helgenom-sekvensering (WGS)81,82,83.
Obs: Det finns en mängd metoder för kvantifiering på - och off-target genomet redigeringar, förklaras ytterligare i olika granskning artiklarna84,85,86.
Representative Results
Dessa experiment Visa hur förmonterade Cas9 RNP kan användas för att manipulera genomen hos primära celler och hela organismer. Forskare renar eller köpa Cas9 protein och sgRNA, kombinera de två komponenterna före bildar komplexet och införa RNP i deras celler eller organismen av intresse. Efter att tillräckligt med tid för redigering för att uppstå och för avkomma av nästa generation att födas (i tillämpliga fall), kontrollera om fenotyper och/eller samla in celler för genotypning. Fenotyper kan observeras via funktionella analyser, uttryck analyser, visualisering (av ögat eller med mikroskopi) eller andra metoder, beroende på experimentet.
Exempelvis kan HSPCs som har redigerats för att korrigera β-globin mutationen som orsakar sicklecellanemi differentieras efter erytrocyter och analyseras för produktionen av friska eller skäran hemoglobin27,87 (figur 1 A). T-cellerna redigeras för att slå ut den hög affinity IL-2 receptorn gen, CD25 (IL2RA), kan analyseras av surface färgning och flöde flödescytometri88, och funktionellt analyseras för att upptäcka en signalering svar på IL-2 stimulering (figur 1B ). T-celler kan också programmeras i många kliniskt viktigt sätt som kräver bedömning av olika fenotyper, inklusive effekten av HIV infektion89 och i vivo antitumöreffekt effekten av bil-T celler11.
Med en co-CRISPR/co-conversion screening metod, redigeras C. elegans maskar samtidigt på två loci62. HDR på Referensgenen dpy-10 med hjälp av en ssODN reparera mallresultat i ett enkelt brytskåra dominerande dpy-10 gain-of-function mutation. Heterozygota F1dpy-10(gof) djur är rullen (Rol) och homozygot dpy-10(gof) djur är dumpy (Dpy). Förekomsten av fenotypen indikerar att Cas9 redigering inträffade i dessa djur och förbättrar oddsen för att identifiera en redigering händelse på det andra locus i Rol eller Dpy F1 djuren. Ett framgångsrikt redigering experiment bör resultera i 33-50% av injicerad P0 maskar ger 20 eller fler F1 avkomma som är Rol eller Dpy90. Det är då möjligt att välja icke-Rol djur att återvända dpy-10 till vildtyp och välj för homozygot redigera sevärdheter. Som en tumregel bör koncentrationen av den crRNA inriktning co-CRISPR Referensgenen hälften att av crRNA riktade mot gen av intresse. Om en redigering i genen av intresse inte återvinns, kan var kvoterna för de två CRISPR-RNAs justeras för att öka sannolikheten för att återställa den önskade mutationen. Exempelvis ökar ökar mängden crRNA för gen av intresse i förhållande till referens gen crRNA andelen maskar som har redigeringar i genen av intresse inom befolkningen av maskar som också äger redigeringar på referens gen locus. Samtidig konvertering frekvenser varierar, men priserna är oftast 20-60%, vilket ofta ger homozygot redigeringar i F1 generationen (figur 1C).
P. hawaiensis ungar som har redigerats för att slå ut den Buksmärta-B -genen (Abd-B) Visa tydliga morfologiska abnormiteter3 (figur 1D). Denna gen krävs för korrekt buk mallning och dess störningar resulterar i bröstkorg-typ hoppa och promenader ben ersätter simning och ankare benen som vanligtvis presentera på buken.
Fastställande av genomet redigering utfall på genotypiska nivå kräver antingen sekvensering eller ett in vitro- test som upptäcker sekvens ändringar. Här visar vi representativa sekvensering data från vår modell celltyper och organismer, belysa olika sätt att redigera kvantifiering. Observera att figur etiketterna är generell eftersom alla metoder som visas här kan tillämpas på alla biologiska system.
Sekvensering tillvägagångssätt varierar i tekniska komplexitet och djup av resultat. För klonal redigerade populationer eller lätta att separera enskilda organismer, kan redigerade individer sekvenseras efter genomisk DNA-extraktion. Standard Sanger sekvensering resultat kommer att avslöja sekvens förändringen på webbplatsen Cas9-cut i en viss individ, med hypotetiska bassubstitutioner som skulle störa dess funktion (figur 2A). Online-verktyget används för sekvensering är en annan Sanger sekvensering-baserad metod som kan tillämpas på blandade populationer snarare än enskilda mutanter78. Sekvenser analyseras med ett online verktyg som kan approximera redigering totaleffektiviteten samt dominerande sekvens utfall. De representativa uppgifterna visas i figur 2B.
Den mest grundliga sekvensering metod som beskrivs här är djupsekvensering (ibland benämnd hög genomströmning eller nästa generations sekvensering). Denna metod ger DNA-sekvenser från enskilda arvsmassan i en blandad befolkning. Sådana uppgifter kan illustreras i en mängd olika sätt. Här har vi klassificerat enskilda sekvensering läsningar från redigerade celler baserat på redigering resultatet (figur 2C). De flesta celler redigeras via den NHEJ vägen, vilket oftast resulterar i gen störningar. I andra har målgenen bytts ut för en alternativ version via HDR27.
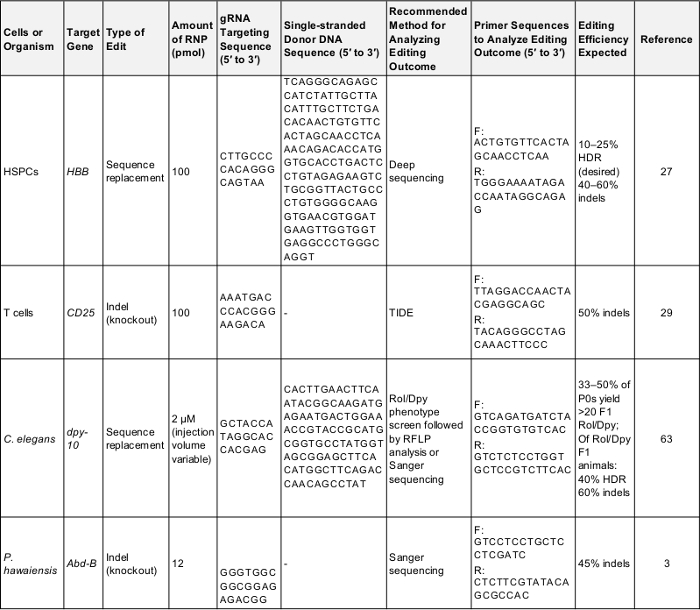
Tabell 1: positiva kontroller för preliminära genomet redigering experiment. Tabellen visar den viktigaste informationen som behövs för att utföra en första gången genomet redigering experiment i alla celler och organismer som beskrivs i detta protokoll. Efter dessa parametrar kan ge ett lyckat resultat som kan användas för att testa protokollet eller som utgångspunkt för jämförelse en gång experimenter riktar en gen av eget intresse. F: framåt, R: bakåt, HDR: homologi-regisserad reparation. Vänligen klicka här för att hämta tabellen.

Figur 1 : Representant fenotypiska resultat från Cas9 RNP redigering av primära mänskliga celler och organismer. (A) detta är en HPLC spår visar att efter framgångsrika genomet redigering, HSPCs som göras åtskillnad mellan till sent stadium erytroblaster kommer att producera mer funktionell hemoglobin än skäran hemoglobin. Mutant erytrocyter producera skäran hemoglobin (HbS), medan framgångsrikt-redigerade celler kommer att producera friska hemoglobin (HbA och HbA2) samt fetalt hemoglobin (HbF). Absorbansen är diagram i godtyckliga enheter (au). Denna panel publicerades först i DeWitt et al. 27. det återges med tillstånd från American Association för befordran av vetenskap. (B) till vänster, för varje tillstånd, i denna panel visas flödescytometri flödesdata visar att ytan-färgade T-celler inte uttrycker CD25 efter CD25 genen har varit utslagen med RNP. CD25 överflödet ritas på x-axeln med Cellstorlek på y-axeln. I denna panel visas till höger, för varje tillstånd, Phospho-Stat5 (pStat5) kvantifiering efter induktion med IL-2. Den signalering reduceras när IL-2 receptorn är frånvarande (CD25 KO). PStat5 överflödet ritas på x-axeln och de uppgifter som härrör från tre olika nivåer av IL-2 input jämförs vertikalt. (C) i denna panel visas en Caenorhabditis elegans co-CRISPR/co-conversion skärm inriktning dpy-10 som den samtidig konvertering markören. Två guida RNAs mål två loci, dpy-10 och din favorit gen (yfg), i samma P0-injicerade djur. HDR på dpy-10 resulterar en Rol eller Dpy fenotyp. Valet av Rol - eller Dpy-F1 djur ökar chanserna för att identifiera redigeringar på det andra locus. (D) denna panel visar att vildtyp Parhyale hawaiensis ungar har normala magar med simning och ankare ben. De Abd-B knock-out ungarna (F0 individer) utveckla en buken omvandlas mot bröstkorgen. Således är det simning och ankare ben borta och ersättas med hoppande och promenader benen är associerad med en normal bröstkorg. Klicka här för att se en större version av denna siffra.

Figur 2 : Typiska resultat från redigering resultatet analysmetoder. (A) i denna panel visas exempel på Sanger sekvensering resultat från enskilda F1 P. hawaiensis organismer, inklusive sekvensen vildtyp och tre olika indels som stör funktionen genen genom att skifta ramen öppen läsning. (B) dessa TIDE resultaten visar antalet infogningar och borttagning händelser som inträffat på en Cas9-målwebbplats i en pool av sekvenserade T-celler. X-axeln anger längden på en viss infogning eller borttagning i nukleotider. (C) dessa djupsekvensering resultaten visar ingen gen editering utan nucleofection eller gärna och framgångsrika redigering med intakt Cas9 RNP, grupperade efter DNA reparation resultatet i HSPCs. vänligen klicka här för att visa en större version av denna siffra.
Discussion
Att upprätta en robust genomet redigering protokoll i en cell linje eller organismen av intresse kräver optimering och empirisk testning av flera viktiga parametrar, behandlas i detta avsnitt. Försöker några varianter av de allmänna strategier som presenteras här är mycket uppmuntras. Den viktigaste begränsningen av detta protokoll är att tillämpa dessa metoder till andra celler eller organismer kan leda till ett annat resultat beroende på vilken art som studerade och en experimentell design som leder till en högeffektiv gen knockout får inte marknadsföra DNA införande. Således rekommenderar vi börjar med de metoder som presenteras här och felsökning som beskrivs nedan.
Felsökning genomet redigering reagens kvalitet:
Generera eller köpa högkvalitativa reagenser är ett kritiskt steg i någon genomet redigering protokoll. Cas9 protein kan renas i labbet eller köpas kommersiellt. Många protokoll Observera en slutlig koncentration för Cas9 i RNP recept, men optimal genen redigering aktivitet beror på den specifika aktiviteten av någon enskild Cas9 protein beredning, som varierar beroende på källan. När protokollet presenteras här arbetar, överväga att optimera mängden RNP används av titrering Cas9 nivåer för att fastställa en optimal koncentration: en som ger mycket specifik DNA klyvning utan onödiga off-target klyvning orsakas av överdriven Cas940.
Guide RNA renhet och homogenitet kan också vara faktorer som påverkar genomet redigering framgång22. Köpta sgRNAs eller separat crRNA och tracrRNA komponenter är generellt högkvalitativa reagenser och en mängd kemiska modifieringar finns att bekämpa problemen med RNA nedbrytning eller att genomsyra ytterligare funktioner till RNP91. Även kemiskt modifierade gRNAs inte kanske är nödvändiga för standarden genomet redigering experiment, vissa grupper har observerat mycket högre redigering effektivitetsvinster med sådant reagens, så att de kan vara värt att försöka efter mastering processen och/eller när gärna nedbrytning verkar vara en fråga22,91. In vitro transkription och efterföljande gel rening är ett billigt alternativ, som kan vara tillräckligt för rutinmässig genomet redigering experiment17,21,49,50. Dessutom flera metoder som används vanligtvis för att producera homogen gärna populationer i vivo, inklusive ribozym - och tRNA-baserade excision av enskilda guider, kan förlängas till in vitro- RNA förberedelse att generera renare produkter92.
Guide RNA och givare DNA design tips:
Guide RNA urval är en kritisk faktor för att uppnå mycket effektiv på målet redigering samtidigt minimera chanserna för off-target klyvning. För att underlätta guide urval, har flera studier använt hög genomströmning skärmar tillsammans med nästa generations sekvensering för att sammanställa sekvens funktioner av framgångsrika guider47,79,93,94, 95,96. Dessa funktioner har använts för att utveckla förutsägande algoritmer och online verktyg för att bistå i guide urval44,45,46,47,48. Sådana algoritmer är jordad på skärmar med DNA-baserade system för guide RNA uttryck. Guider uttrycks med hjälp av en Pol III arrangören, och deras uttryck är därför benägen att de begränsningar som är associerade med Pol III transkription, såsom förtida upphörande när de stöter på spår av uracil97,98, 99. Användning av RNPs gjorde dock med in vitro-synthesized guide RNAs kringgår dessa farhågor och förenklar begränsningarna som guide-design. En gemensam funktion som vuxit fram ur dessa algoritmer och har bekräftats i ett flertal studier med mycket effektiv gen editering, är förekomsten av en purin, särskilt en guanin, 3′ slutet av guidens mål-specifika sekvens. Denna guide funktion har varit mycket framgångsrikt bland organismer alltifrån däggdjur till C. elegans, bananflugor och zebrafiskar65,100,101. Dessutom för C. elegansär utforma guider med en GG-dinukleotid 3′ slutet av guidens inriktning regionen en effektiv strategi för att förutsäga mycket effektiv guide RNAs65. Idealiskt, testa flera guider parallellt för att avgöra vilken som är mest framgångsrika för ett visst program.
När du försöker införa en DNA-sekvens i arvsmassan, är utformningen av den givaren eller templat-DNA också avgörande. Enkelsträngat oligonukleotiden givare (ssODNs) infogas mer tillförlitligt än andra typiska reparation mallar, linjärt dubbelsträngat och plasmid DNA54,55,102. På vissa lokus, kan HDR effektivitet förbättras med ssODNs som kompletterar målart eller fördrivna DNA-strängen och besitter homologi armar som är asymmetriska i längd27,55. Eftersom mallen reparation infogas på webbplatsen skär och innehåller sekvensen riktade, måste åtgärder vidtas för att förhindra Cas9 klyva donatorns DNA före eller efter genomisk insättningspunkten. Detta åstadkoms genom att göra tysta mutationer till PAM sekvens eller utsäde regionen, undvika erkännande av Cas9 samtidigt behålla funktionen av den infogade gen21,103. Om ens enda nukleotid ändringar till PAM är sannolikt att avskaffa bindande104, prova att ändra minst fyra nukleotider för att vara säker.
Betydelse och framtida tillämpningar:
Genomet redigering med CRISPR-Cas9 har vuxit fram som en kraftfull metod som gör det möjligt för facile genmanipulation av någon organism. Redigering med den Cas9 RNP tar lite mer ansträngning först men är enkel att använda när reagenser och protokoll är etablerade i ett labb. Redigera celler med förmonterade RNP istället för plasmid DNA leder till högre övergripande redigering effektivitetsvinster, inklusive svåra att nå gen insättningspunkten via HDR, med färre off-target effekter24,25,26 , 27 , 29. vidare praktiker undvika problem med genuttryck RNA nedbrytning, proteinveckning och associationen mellan gärna och Cas9 molekyler syntetiseras separat inom den cell22,23. RNP redigering också kringgår säkerhet oro för insertionsmutationer och ihållande uttryck som kan uppstå när viral leveransmetoder är används kliniskt14. På grund av dessa fördelar ynnest många forskare bedriver preklinisk, proof-of-concept experiment RNP redigering för mänskliga terapeutiska tillämpningar. Både i vivo och ex vivo RNP-baserade genomet redigering metoder är under utveckling att behandla eller även bota en mängd tillstånd, från genetiska sjukdomar som Duchennes muskeldystrofi105 och sicklecellsjukdom27 till HIV29 och cancer11. Intressant, används Cas9 RNP alltmer som en leveransmetod för jordbrukets teknik eftersom det möjliggör 'DNA-fri' redigering av växter33,34,36.
Disclosures
Författarna Alexander Marson och Jacob E. Corn är grundarna av Spotlight Therapeutics. Jacob E. Corn är rådgivare åt Mission Therapeutics och hans laboratorium har fått sponsrade forskningsstöd från AstraZeneca och Pfizer. Alexander Marson är rådgivare åt Juno Therapeutics och pakten Therapeutics och hans laboratorium har fått sponsrade forskningsstöd från Juno Therapeutics, Epinomics och Sanofi. Hans laboratorium har också ansökt om patent avseende Cas9 RNP-tekniken.
Acknowledgments
Vi tackar många tidigare medlemmar i våra labb och Bay Area genomet redigering gemenskapen för deras bidrag till utvecklingen av dessa metoder. Vi tackar Ross Wilson för att kritiskt läsa detta manuskript.
Alexander Marsons forskning stöds av en gåva från den Jake Aronov och en nationell multipelskleros samfund bevilja (CA 1074-A-21). Alexander Marson rymmer en karriär Award för medicinska forskare från Burroughs Wellcome fonden och är en Chan Zuckerberg Biohub utredare. Jacob E. Corns forskning stöds av Li Ka Shing Foundation, Heritage medicinsk medicinska forskningsinstitut och California Institute for Regenerative Medicine. Behnom Farboud och Barbara J. Meyers forskning finansieras delvis av NIGMS bidraget R01 GM030702 till Barbara J. Meyer, som är en utredare för Howard Hughes Medical Institute. Erin Jarvis och Nipam H. Patels forskning finansieras delvis av NSF bidraget IOS-1257379 och Erin Jarvis erkänner stöd från en NSF-GRFP och en Philomathia Graduate Fellowship.
Materials
| Name | Company | Catalog Number | Comments |
| Reagents/Materials | |||
| DNA oligonucleotides | Integrated DNA Technologies | - | IDT will provide custom DNA sequences, including those in Table 1 |
| Guide RNAs | Synthego | - | Synthego will provide high-quality sgRNAs for S. pyogenes Cas9, including custom sgRNAs containing the targeting sequences included in Table 1 |
| Purified Cas9 protein (EnGen Cas9 NLS, S. pyogenes) | New England Biosciences | M0646T | If possible, purifying Cas9 in-house or purchasing from local core facilities is a less expensive option |
| Normal peripheral blood CD34+ stem/progenitor cells | AllCells | PB032-2 | |
| StemSpan SFEM | StemCell Technologies | 09650 | |
| StemSpan CC110 | StemCell Technologies | 02697 | |
| P3 Primary Cell 4D-Nucleofector X Kit | Lonza | V4XP-3032 | |
| RPMI-1640 Medium, With sodium bicarbonate, without L-glutamine, liquid | Sigma | R0883-6X500ML | |
| EasySep™ Human T Cell Isolation Kit | Stemcell | 17951 | |
| cell culture plate, 96 wells, round | Fisher Scientific | 3799 | |
| CTS (Cell Therapy Systems) Dynabeads CD3/CD28 | Life Tech | 40203D | |
| Reombinant Human IL-2 | UCSF Pharmacy | NA | |
| SepMate-50 500-pack IVD | Stemcell Technologies | 85460 | |
| OP50 Escherichia coli | Caenorhabditis Genetics Center | OP-50 | https://cgc.umn.edu/ |
| Nematode Growth Media agar in petri dishes | - | - | See Stiernagle, T (ref. 59) |
| Standard borosilicate glass capillaries with filament: 4 in (100 mm), 1/0.58 OD/ID | World Precision Instruments | 1B100F-4 | |
| Single-barrel standard borosilicate glass capillaries: 6 in (152 mm), 2/1.12 OD/ID | World Precision Instruments | 1B200-6 | |
| Cover glass; 24 × 50 mm | Thermo Fisher Scientific | 12-544E | |
| Cover glass; 22 × 22 mm | Thermo Fisher Scientific | 12-518-105K | |
| Apex LE agarose | Genesee Scientific | 20-102 | |
| Halocarbon oil 700 | Sigma-Aldrich | H8898-100ML | |
| pCFJ90 plasmid | Addgene | 19327 | |
| Compressed nitrogen | - | ||
| 60 mM culture dishes | BD | ||
| Capillary tubes with filament: 4 in (1.0 mm) | World Precision Instruments | T2100F-4 | |
| Sylgard 184 | Dow Corning | ||
| Petri dishes (100 × 15 mm) | - | ||
| Tungsten wire (0.005 in. diameter) | Ted Pella | ||
| Perfluoroalkoxy alkane (PFA) | - | ||
| Marine salt | - | ||
| 9" pasteur pipettes | - | ||
| Phenol red | - | ||
| Nuclease-free water | - | ||
| Equipment | |||
| 4D Nucleofector | Lonza | AAF-1002X | |
| MZ75 Stereomicroscope | Leica | Out-of-production. Current model is the M80 Stereomicroscope | |
| Axio Vert35 inverted phase contrast fluorescent microscope | Zeiss | Out-of-production. Current model is the Axio VertA.1 | |
| Laser-based micropipette puller (for C. elegans protocol) | Sutter Instrument | FG-P2000 | |
| Picoliter Microinjector (for C. elegans protocol) | Warner Instruments | PLI-100A | |
| Three-axis Joystick oil hydraulic micromanipulator | Narishige International | MO-202U | |
| Coarse manipulator | Narishige International | MMN-1 | |
| Micropipette puller (for P. hawaiensis protocol) | Sutter Instrument | P-80/PC | |
| Microinjector (for P. hawaiensis protocol) | Narishige | IM300 | |
| Microloader pipette tips | Eppendorf | 5242956003 | |
| NG-agar |
References
- Komor, A. C., Badran, A. H., Liu, D. R. CRISPR-based technologies for the manipulation of eukaryotic genomes. Cell. , 1-17 (2016).
- Barrangou, R., Horvath, P. A decade of discovery: CRISPR functions and applications. Nature Microbiology. 2, 1-9 (2017).
- Martin, A., Serano, J. M., et al. CRISPR/Cas9 mutagenesis reveals versatile roles of Hox genes in crustacean limb specification and evolution. Current Biology. 26 (1), 14-26 (2016).
- Goldstein, B., King, N. The future of cell biology: emerging model organisms. Trends in Cell Biology. 26 (11), 818-824 (2016).
- Mali, P., Yang, L., et al. RNA-guided human genome engineering via Cas9. Science. 339 (6121), 823-826 (2013).
- Cong, L., Ran, F. A., et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 339 (6121), 819-823 (2013).
- Deltcheva, E., Chylinski, K., et al. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature. 471 (7340), 602-607 (2011).
- Jinek, M., Chylinski, K., Fonfara, I., Hauer, M., Doudna, J. A., Charpentier, E. A Programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 337 (6096), 816-821 (2012).
- Nowak, C. M., Lawson, S., Zerez, M., Bleris, L. Guide RNA engineering for versatile Cas9 functionality. Nucleic Acids Research. 44 (20), 9555-9564 (2016).
- Jiang, W., Cox, D., Zhang, F., Bikard, D., Marraffini, L. A. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nature Biotechnology. , 1-9 (2013).
- Rupp, L. J., Schumann, K., et al. CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Scientific Reports. 7 (1), 737 (2017).
- Graham, D. B., Root, D. E. Resources for the design of CRISPR gene editing experiments. Genome Biology. 16, 260 (2015).
- Wang, H., La Russa, M., Qi, L. S. CRISPR/Cas9 in genome editing and beyond. Annual Review of Biochemistry. 85, 2270 (2016).
- Nelson, C. E., Gersbach, C. A. Engineering delivery vehicles for genome editing. Annual Review of Chemical and Biomolecular Engineering. 7, 637-662 (2016).
- Yin, H., Kauffman, K. J., Anderson, D. G. Delivery technologies for genome editing. Nature Reviews Drug Discovery. 16 (6), 387-399 (2017).
- Zuris, J. A., Thompson, D. B., et al. Cationic lipid-mediated delivery of proteins enables efficient protein-based genome editing in vitro and in vivo. Nature Biotechnology. 33 (1), 73-80 (2015).
- Cho, S. W., Lee, J., Carroll, D., Kim, J. -S., Lee, J. Heritable gene knockout in Caenorhabditis elegans by direct injection of Cas9-sgRNA ribonucleoproteins. Genetics. 195 (3), 1177-1180 (2013).
- Wang, W., Kutny, P. M., et al. Delivery of Cas9 protein into mouse zygotes through a series of electroporation dramatically increases the efficiency of model creation. Journal of Genetics and Genomics. 43 (5), 319-327 (2016).
- Chen, S., Lee, B., Lee, A. Y. -F., Modzelewski, A. J., He, L. Highly efficient mouse genome editing by CRISPR ribonucleoprotein electroporation of zygotes. Journal of Biological Chemistry. 291 (28), 14457-14467 (2016).
- Remy, S., Chenouard, V., et al. Generation of gene-edited rats by delivery of CRISPR/Cas9 protein and donor DNA into intact zygotes using electroporation. Scientific Reports. 7 (1), 16554 (2017).
- DeWitt, M. A., Corn, J. E., Carroll, D. Genome editing via delivery of Cas9 ribonucleoprotein. Methods. , 1-7 (2017).
- Hendel, A., Bak, R. O., et al. Chemically modified guide RNAs enhance CRISPR-Cas genome editing in human primary cells. Nature Biotechnology. 33 (9), 985-989 (2015).
- Thyme, S. B., Akhmetova, L., Montague, T. G., Valen, E., Schier, A. F. Internal guide RNA interactions interfere with Cas9-mediated cleavage. Nature Communications. 7, 11750 (2016).
- Kim, S., Kim, D., Cho, S. W., Kim, J., Kim, J. -S. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Research. 24 (6), 1012-1019 (2014).
- Lin, S., Staahl, B. T., Alla, R. K., Doudna, J. A. Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. eLife. 3, 04766 (2014).
- Liang, X., Potter, J., et al. Rapid and highly efficient mammalian cell engineering via Cas9 protein transfection. Journal of Biotechnology. 208, 44-53 (2015).
- DeWitt, M. A., Magis, W., Bray, N. L., Wang, T. Selection-free genome editing of the sickle mutation in human adult hematopoietic stem/progenitor cells. Science Translational Medicine. 8 (360), (2016).
- Ramakrishna, S., Kwaku Dad, A. -B., Beloor, J., Gopalappa, R., Lee, S. -K., Kim, H. Gene disruption by cell-penetrating peptide-mediated delivery of Cas9 protein and guide RNA. Genome Research. 24 (6), 1020-1027 (2014).
- Schumann, K., Lin, S., et al. Generation of knock-in primary human T cells using Cas9 ribonucleoproteins. Proceedings of the National Academy of Sciences of the United States of America. 112 (33), 10437-10442 (2015).
- Lee, J. -S., Kwak, S. -J., et al. RNA-guided genome editing in Drosophila with the purified Cas9 protein. G3: Genes, Genomes, Genetics (Bethesda, MD). 4 (7), 1291-1295 (2014).
- Sung, Y. H., Kim, J. M., et al. Highly efficient gene knockout in mice and zebrafish with RNA-guided endonucleases. Genome Research. 24 (1), 125-131 (2014).
- Menoret, S., De Cian, A., et al. Homology-directed repair in rodent zygotes using Cas9 and TALEN engineered proteins. Scientific Reports. 5, 14410 (2015).
- Woo, J. W., Kim, J., et al. DNA-free genome editing in plants with preassembled CRISPR-Cas9 ribonucleoproteins. Nature Biotechnology. 33 (11), 1162-1164 (2015).
- Malnoy, M., Viola, R., et al. DNA-free genetically edited grapevine and apple protoplast using CRISPR/Cas9 ribonucleoproteins. Frontiers in Plant Science. 7, 1904 (2016).
- Svitashev, S., Schwartz, C., Lenderts, B., Young, J. K., Mark Cigan, A. Genome editing in maize directed by CRISPR-Cas9 ribonucleoprotein complexes. Nature Communications. 7, 13274 (2016).
- Liang, Z., Chen, K., et al. Efficient DNA-free genome editing of bread wheat using CRISPR/Cas9 ribonucleoprotein complexes. Nature Communications. 8, 14261 (2017).
- Shin, S. -E., Lim, J. -M., et al. CRISPR/Cas9-induced knockout and knock-in mutations in Chlamydomonas reinhardtii. Scientific Reports. 6, 27810 (2016).
- Pohl, C., Kiel, J. A. K. W., Driessen, A. J. M., Bovenberg, R. A. L., Nygård, Y. CRISPR/Cas9 based genome editing of Penicillium chrysogenum. ACS Synthetic Biology. 5 (7), 754-764 (2016).
- Grahl, N., Demers, E. G., Crocker, A. W., Hogan, D. A. Use of RNA-protein complexes for genome editing in non-albicans Candida species. mSphere. 2 (3), (2017).
- Rivera-Torres, N., Kmiec, E. B. A standard methodology to examine on-site mutagenicity as a function of point mutation repair catalyzed by CRISPR/Cas9 and ssODN in human cells. Journal of Visualized Experiments. (126), (2017).
- Nandal, A., Mallon, B., Telugu, B. P. Efficient generation and editing of feeder-free IPSCs from human pancreatic cells using the CRISPR-Cas9 system. Journal of Visualized Experiments. (129), (2017).
- Mohr, S. E., Hu, Y., Ewen-Campen, B., Housden, B. E., Viswanatha, R., Perrimon, N. CRISPR guide RNA design for research applications. The FEBS Journal. 283 (17), 3232-3238 (2016).
- Bauer, D. E., Canver, M. C., Orkin, S. H. Generation of genomic deletions in mammalian cell lines via CRISPR/Cas9. Journal of Visualized Experiments. (95), e52118 (2015).
- Hsu, P. D., Scott, D. A., et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nature Biotechnology. 31 (9), 827-832 (2013).
- Heigwer, F., Kerr, G., Boutros, M. E-CRISP: fast CRISPR target site identification. Nature Methods. 11 (2), 122-123 (2014).
- Moreno-Mateos, M. A., Vejnar, C. E., et al. CRISPRscan: designing highly efficient sgRNAs for CRISPR-Cas9 targeting in vivo. Nature Methods. 12 (10), 982-988 (2015).
- Labun, K., Montague, T. G., Gagnon, J. A., Thyme, S. B., Valen, E. CHOPCHOP v2: a web tool for the next generation of CRISPR genome engineering. Nucleic Acids Research. 44, 272-276 (2016).
- Haeussler, M., Schönig, K., et al. Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biology. 17 (1), 148 (2016).
- Lo, T. -W., Pickle, C. S., et al. Precise and heritable genome editing in evolutionarily diverse nematodes using TALENs and CRISPR/Cas9 to engineer insertions and deletions. Genetics. 195 (2), 331-348 (2013).
- Bassett, A., Liu, J. -L. CRISPR/Cas9 mediated genome engineering in Drosophila. Methods. 69 (2), 128-136 (2014).
- Prior, H., Jawad, A. K., MacConnachie, L., Beg, A. A. Highly efficient, rapid and co-CRISPR independent genome editing in Caenorhabditis elegans. G3: Genes, Genomes, Genetics. , Bethesda, MD. (2017).
- Hirsh, A. Cas9 expression and purification protocol. protocols.io. , (2017).
- DeWitt, M. A., Wong, J. In vitro transcription of guide RNAs. protocols.io. , (2017).
- Yang, L., Guell, M., et al. Optimization of scarless human stem cell genome editing. Nucleic Acids Research. 41 (19), 9049-9061 (2013).
- Richardson, C. D., Ray, G. J., DeWitt, M. A., Curie, G. L., Corn, J. E. Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA. Nature Biotechnology. 34 (3), 339-344 (2016).
- Paix, A., Folkmann, A., Seydoux, G. Precision genome editing using CRISPR-Cas9 and linear repair templates in C. elegans. Methods. 121-122, 86-93 (2017).
- Mello, C., Fire, A. DNA transformation. Methods in Cell Biology. 48, 451-482 (1995).
- Sutter Pipette Cookbook. , Available from: https://www.sutter.com/PDFs/pipette_cookbook.pdf (2017).
- Stiernagle, T. Maintenance of C. elegans. WormBook: the online review of C. elegans biology. , (2006).
- Evans, T. C. Transformation and microinjection. WormBook: the online review of C. elegans biology. , (2006).
- Berkowitz, L. A., Knight, A. L., Caldwell, G. A., Caldwell, K. A. Generation of stable transgenic C. elegans using microinjection. Journal of Visualized Experiments. (18), (2008).
- Kim, H., Ishidate, T., et al. A co-CRISPR strategy for efficient genome editing in Caenorhabditis elegans. Genetics. 197 (4), 1069-1080 (2014).
- Arribere, J. A., Bell, R. T., Fu, B. X. H., Artiles, K. L., Hartman, P. S., Fire, A. Z. Efficient marker-free recovery of custom genetic modifications with CRISPR/Cas9 in Caenorhabditis elegans. Genetics. 198 (3), 837-846 (2014).
- Ward, J. D. Rapid and precise engineering of the Caenorhabditis elegans genome with lethal mutation co-conversion and inactivation of NHEJ repair. Genetics. 199 (2), 363-377 (2015).
- Farboud, B., Meyer, B. J. Dramatic enhancement of genome editing by CRISPR/Cas9 through improved guide RNA design. Genetics. 199 (4), 959-971 (2015).
- Wood, A. J., Lo, T. -W., et al. Targeted genome editing across species using ZFNs and TALENs. Science. 333 (6040), 307 (2011).
- Friedland, A. E., Tzur, Y. B., Esvelt, K. M., Colaiácovo, M. P., Church, G. M., Calarco, J. A. Heritable genome editing in C. elegans via a CRISPR-Cas9 system. Nature Methods. 10 (8), 741-743 (2013).
- Dickinson, D. J., Ward, J. D., Reiner, D. J., Goldstein, B. Engineering the Caenorhabditis elegans genome using Cas9-triggered homologous recombination. Nature Methods. 10 (10), 1028-1034 (2013).
- Rehm, E. J., Hannibal, R. L., Chaw, R. C., Vargas-Vila, M. A., Patel, N. H. Injection of Parhyale hawaiensis blastomeres with fluorescently labeled tracers. Cold Spring Harbor Protocols. 2009 (1), (2009).
- Gerberding, M., Browne, W. E., Patel, N. H. Cell lineage analysis of the amphipod crustacean Parhyale hawaiensis reveals an early restriction of cell fates. Development (Cambridge, England). 129 (24), 5789-5801 (2002).
- Browne, W. E., Price, A. L., Gerberding, M., Patel, N. H. Stages of embryonic development in the amphipod crustacean, Parhyale hawaiensis. Genesis. 42 (3), New York, NY. 124-149 (2005).
- Rehm, E. J., Hannibal, R. L., Chaw, R. C., Vargas-Vila, M. A., Patel, N. H. Fixation and dissection of Parhyale hawaiensis embryos. Cold Spring Harbor Protocols. 2009 (1), (2009).
- Rehm, E. J., Hannibal, R. L., Chaw, R. C., Vargas-Vila, M. A., Patel, N. H. In situ hybridization of labeled RNA probes to fixed Parhyale hawaiensis embryos. Cold Spring Harbor Protocols. 2009 (1), (2009).
- Rehm, E. J., Hannibal, R. L., Chaw, R. C., Vargas-Vila, M. A., Patel, N. H. Antibody staining of Parhyale hawaiensis embryos. Cold Spring Harbor Protocols. 2009 (1), (2009).
- Kontarakis, Z., Pavlopoulos, A. Transgenesis in non-model organisms: the case of Parhyale. Methods in Molecular Biology. 1196, Clifton, NJ. 145-181 (2014).
- Kim, H. J., Lee, H. J., Kim, H., Cho, S. W., Kim, J. -S. Targeted genome editing in human cells with zinc finger nucleases constructed via modular assembly. Genome Research. 19 (7), 1279-1288 (2009).
- Qiu, P., Shandilya, H., D'Alessio, J. M., O'Connor, K., Durocher, J., Gerard, G. F. Mutation detection using Surveyor nuclease. BioTechniques. 36 (4), 702-707 (2004).
- Brinkman, E. K., Chen, T., Amendola, M., van Steensel, B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Research. 42 (22), 168 (2014).
- Tsai, S. Q., Zheng, Z., et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nature Biotechnology. 33 (2), 187-197 (2015).
- Frock, R. L., Hu, J., Meyers, R. M., Ho, Y. -J., Kii, E., Alt, F. W. Genome-wide detection of DNA double-stranded breaks induced by engineered nucleases. Nature Biotechnology. 33 (2), 179-186 (2015).
- Smith, C., Gore, A., et al. Whole-genome sequencing analysis reveals high specificity of CRISPR/Cas9 and TALEN-based genome editing in human iPSCs. Cell Stem Cell. 15 (1), 12-13 (2014).
- Veres, A., Gosis, B. S., et al. Low incidence of off-target mutations in individual CRISPR-Cas9 and TALEN targeted human stem cell clones detected by whole-genome sequencing. Cell Stem Cell. 15 (1), 27-30 (2014).
- Kim, D., Bae, S., et al. Digenome-seq: genome-wide profiling of CRISPR-Cas9 off-target effects in human cells. Nature Methods. 12 (3), 237-243 (2015).
- Hendel, A., Fine, E. J., Bao, G., Porteus, M. H. Quantifying on- and off-target genome editing. Trends in Biotechnology. 33 (2), 132-140 (2015).
- O'Geen, H., Yu, A. S., Segal, D. J. How specific is CRISPR/Cas9 really. Current Opinion in Chemical Biology. 29, 72-78 (2015).
- Tsai, S. Q., Joung, J. K. Defining and improving the genome-wide specificities of CRISPR-Cas9 nucleases. Nature Reviews Genetics. 17 (5), 300-312 (2016).
- Hoban, M. D., Cost, G. J., et al. Correction of the sickle cell disease mutation in human hematopoietic stem/progenitor cells. Blood. 125 (17), 2597-2604 (2015).
- Simeonov, D. R., Gowen, B. G., et al. Discovery of stimulation-responsive immune enhancers with CRISPR activation. Nature. , (2017).
- Hultquist, J. F., Schumann, K., et al. A Cas9 ribonucleoprotein platform for functional genetic studies of HIV-host interactions in primary human T cells. Cell Reports. 17 (5), 1438-1452 (2016).
- Paix, A., Wang, Y., et al. Scalable and versatile genome editing using linear DNAs with microhomology to Cas9 sites in Caenorhabditis elegans. Genetics. 198 (4), 1347-1356 (2014).
- Lee, K., Mackley, V. A., et al. Synthetically modified guide RNA and donor DNA are a versatile platform for CRISPR-Cas9 engineering. eLife. 6, (2017).
- Minkenberg, B., Wheatley, M., Yang, Y. CRISPR/Cas9-enabled multiplex genome editing and its application. Progress in Molecular Biology and Translational Science. 149, 111-132 (2017).
- Doench, J. G., Fusi, N., et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nature Biotechnology. 34 (2), 184-191 (2016).
- Doench, J. G., Hartenian, E., et al. Rational design of highly active sgRNAs for CRISPR-Cas9-mediated gene inactivation. Nature Biotechnology. , 1-8 (2014).
- Liu, H., Wei, Z., Dominguez, A., Li, Y., Wang, X., Qi, L. S. CRISPR-ERA: a comprehensive design tool for CRISPR-mediated gene editing, repression and activation. Bioinformatics (Oxford, England). 31 (22), 3676-3678 (2015).
- Wu, X., Scott, D. A., et al. Genome-wide binding of the CRISPR endonuclease Cas9 in mammalian cells. Nature Biotechnology. 32 (7), 670-676 (2014).
- Bogenhagen, D. F., Brown, D. D. Nucleotide sequences in Xenopus 5S DNA required for transcription termination. Cell. 24 (1), 261-270 (1981).
- Cozzarelli, N. R., Gerrard, S. P., Schlissel, M., Brown, D. D., Bogenhagen, D. F. Purified RNA polymerase III accurately and efficiently terminates transcription of 5S RNA genes. Cell. 34 (3), 829-835 (1983).
- Chen, B., Gilbert, L. A., et al. Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell. 155 (7), 1479-1491 (2013).
- Gagnon, J. A., Valen, E., et al. Efficient mutagenesis by Cas9 protein-mediated oligonucleotide insertion and large-scale assessment of single-guide RNAs. PLoS ONE. 9 (5), 98186 (2014).
- Ren, X., Yang, Z., et al. Enhanced specificity and efficiency of the CRISPR/Cas9 system with optimized sgRNA parameters in Drosophila. Cell Reports. 9 (3), 1151-1162 (2014).
- Ran, F. A., Hsu, P. D., Wright, J., Agarwala, V., Scott, D. A., Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nature Protocols. 8 (11), 2281-2308 (2013).
- Serano, J. M., Martin, A., et al. Comprehensive analysis of Hox gene expression in the amphipod crustacean Parhyale hawaiensis. Developmental Biology. 409 (1), 297-309 (2016).
- Sternberg, S. H., Redding, S., Jinek, M., Greene, E. C., Doudna, J. A. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature. , 1-17 (2014).
- Lee, K., Conboy, M., et al. Nanoparticle delivery of Cas9 ribonucleoprotein and donor DNA in vivo induces homology-directed DNA repair. Nature Biomedical Engineering. 1 (11), 889-901 (2017).
