

Summary
Utilisant un Cas9 préassemblé complexe ribonucléoprotéique (RNP) est une méthode puissante pour l’édition de génome précis et efficace. Ici, nous mettons en évidence son utilité dans un large éventail de cellules et des organismes, y compris des cellules humaines primaires et les deux classiques et de nouveaux organismes modèles.
Abstract
Édition de génome eucaryote in situ avec CRISPR (cluster régulièrement dois‑je courtes palindromes répétitions)-systèmes de Cas (CRISPR-associés) est rapidement devenu un lieu commun entre chercheurs poursuivant une grande variété de questions biologiques. Les utilisateurs emploient plus souvent la protéine Cas9 dérivée de Streptococcus pyogenes dans un complexe avec un guide facilement reprogrammé RNA (gRNA). Ces composants sont introduits dans les cellules, et à travers une base formant un couple avec une région complémentaire du génome (dsDNA) l’ADN double-brin, l’enzyme s’attache les deux brins pour générer une interruption bicaténaires (DSB). Réparation ultérieure mène à insertion aléatoire ou délétions (indels) ou l’incorporation d’ADN fournis par l’expérimentateur sur le site de la pause.
L’utilisation d’un unique-guide ARN et Cas9 protéine purifiée, prémonté pour former un RNP et envoyées directement aux cellules, est une approche puissante pour la réalisation de montage de gènes très efficace. RNP édition particulièrement augmente la vitesse d’insertion de gènes, un résultat qui est souvent difficile à atteindre. Par rapport à la livraison via un plasmide, la persistance plus courte de la RNP Cas9 au sein de la cellule conduit à moins d’événements hors-cible.
Malgré ses avantages, beaucoup d’utilisateurs occasionnels de l’édition de gène CRISPR est moins familiers avec cette technique. Pour abaisser la barrière à l’entrée, nous exposons des protocoles détaillés pour la mise en œuvre de la stratégie RNP dans des situations différentes, mettant en évidence ses avantages distincts et diverses applications. Nous couvrons édition dans deux types de cellules humaines primaires, les lymphocytes T et les cellules souches/progénitrices hématopoïétiques (HSPCs). Nous montrons aussi comment Cas9 RNP montage permet la facile manipulation génétique des organismes entiers, y compris l’ascaride lombricoïde le classique modèle Caenorhabditis elegans et plus récemment introduit crustacé modèle, Parhyale affinis.
Introduction
FLE CRISPR-Cas9 système permet aux scientifiques d’altérer les régions ciblées de n’importe quel génome1. Cette technique rapide et peu coûteuse a révolutionné la recherche fondamentale et promet de faire un impact profond sur le développement de thérapies pour les maladies personnalisé, agriculture de précision et au-delà de2. CRISPR édition est un outil de démocratisation et application du régime dans un nouveau laboratoire ne nécessite aucune expertise particulière dans les compétences de génome engineering, base de la biologie moléculaire. Maintenant, les chercheurs peuvent étudier les organismes auparavant insolubles avec quelques variantes pour les manipulations génétiques3,4. Au cours des cinq dernières années, les CRISPR génome édition a servi à plus de 200 différents vertébrés, invertébrés, plantes et espèces microbiennes de l’ingénieur.
Adapté de la voie de la défense procaryotes CRISPR, les éléments de base requis pour la modification du génome in situ sont la protéine de Cas9, généralement à partir de S. pyogenes et codon optimisé avec un signal de localisation nucléaire supplémentaire (NLS) et ses spécialisées Guide de RNA5,6. Si ne pas discuté ici, autres Cas9 orthologues ou endonucléases CRISPR peuvent également être utilisés. La gRNA naturelle est composée de deux pièces séparément transcrits, l’ARN CRISPR (crRNA) et l’activation de trans-crRNA (tracrRNA)7. Ces ARN peut être fusionnés en une seule transcription, connue comme le single-guide RNA (sgRNA)8. La plupart des éditeurs de génome choisissent la simplifiée sgRNA9, bien que le double-guide est aussi régulièrement utilisé10,11. Expérimentateurs choisissent une cible de l’ADN génomique 20 nucléotides (nt), veiller à ce qu’il se trouve à côté d’une courte signature licences requise pour la reconnaissance de Cas9, appelée un motif adjacent de protospacer (PAM) et conçoivent un gRNA qui contient la séquence complémentaire12 .
Une fois à l’intérieur de la cellule, le RNP complexe localise sa cible génomique, les paires de bases gRNA avec l’ADN complémentaire chapelet, et puis l’enzyme s’attache les deux brins d’ADN pour générer un double brin cassent2. Machines de réparation cellulaire fixe l’ORD par l’une des deux voies au moins : par la voie de (NHEJ) fin-joining erreurs non homologue ou la réparation d’homologie dirigée (HDR), qui intègre parfaitement ADN contenant « des armes » d’homologie de part et d’autre de la pause. La voie de réparation ancienne mène habituellement à la formation indel et désorganisation du gène qui en découle, alors que ce dernier permet aux expérimentateurs insérer ou modifier de séquences ADN1.
L’efficacité et la précision édition dépendent des moyens par lequel Cas9 et gRNA entrent dans la cellule. Ces composants peuvent être livrés aux cellules cultivées, des embryons ou des organismes sous forme d’acides nucléiques ou comme un préassemblé RNP complexe13,14,15. Les méthodes courantes de livraison à base d’acide nucléique sont la transduction virale, transfection ou électroporation des ARNm ou l’ADN de plasmide. Guide RNA et Cas9 protéines sont ensuite produits au sein de la cellule et ils s’associent pour former un complexe.
La livraison directe du RNP exige la purification distincte du guide RNA et de protéines de Cas9. Cela peut être fait en interne, ou les protéines et les sgRNA peuvent être achetés de l’un de plusieurs fournisseurs commerciaux. Une fois acquis, le Cas9 gRNA mélangés pour former le complexe enzymatiquement compétent de RNP et sont introduits aux cellules par injection directe dans les œufs/embryons fertilisés, transfection à base de lipides16ou électroporation. Le premier rapport du RNP montage injection impliquée dans c. elegans gonades17. Microinjection est encore le moyen privilégié d’introduire les RNP des embryons et des organismes entiers, bien qu’efficace électroporation a été démontrée en18,19 et rat20 des embryons de souris. Nous décrivons des protocoles permettant d’injecter directement des RNP dans les gonades de c. elegans et d’embryons P. affinis et recommander un type spécialisé de l’électroporation pour livrer RNP lors de la modification des cellules humaines primaires. Cette méthode, nucleofection, implique des programmes optimisés électroporation et solutions spécifiques à un type de cellule et permet le RNP à entrer le cytoplasme et le noyau21.
L’édition de génome avec RNP propose plusieurs avantages distincts. Parce que les protéines et les ARN est préassemblées et qualité peut être assurée avant la livraison, RNP édition évite les nombreux pièges liés à la prestation de base d’acide nucléique. À savoir, il n’y a aucun risque d’intégration Cas9-codage ADN dans le génome hôte, ARNm n’est jamais exposée pour dégradation et elle contourne les problèmes avec in vivo gRNA ou protein expression, pliage et association22,23. En outre, à l’aide de RNP mène pour réduire la toxicité et beaucoup moins d’événements hors-cible que l’expression axée sur le plasmide, un résultat de demi-vie plus courte de la RNP à l’intérieur de la cellule24,25,26,27.
Enfin, RNP édition manifestement conduit à des taux élevés de montage dans une variété de lignées cellulaires humaines, cellules primaires tels que les fibroblastes, cellules souches embryonnaires (CSE), induite par les cellules souches pluripotentes (ISPC), HSPCs, et les cellules T16,24, 25,26,27,28,29; chez les invertébrés, notamment c. elegans, P. affiniset drosophiles3,17,30; en espèces de vertébrés comme le poisson-zèbre, des souris et des rats31,32; à planter des essences dont Arabidopsis, tabac, laitue, riz, vigne, pomme, maïs et blé33,34,35,36; et dans Chlamydomonas, Penicilliumet Candida espèces37,38,39. La fréquence de formation indel peut être plus élevée lorsque vous utilisez RNP par rapport à la prestation de plasmide, et insertion de l’ADN induite par le HDR peut être plus facile d’atteindre25,27,29.
Le protocole décrit ici utilise le RNP Cas9 et est une technique efficace, aisément adaptable qui est simple à appliquer à une grande variété de40,des systèmes biologiques41, en particulier dans les cellules qui sont par ailleurs difficiles à travailler et dans les organismes dépourvus de systèmes bien établis pour les manipulations génétiques précises. Nous commençons par décrire comment concevoir et assembler le RNP Cas9 avant de couvrir son utilisation à travers des organismes et des types de cellules différents modèle obtenir. Les cellules souches/progénitrices hématopoïétiques (HSPCs) et les lymphocytes T sont édités en utilisant la même méthode, nucleofection, afin qu’ils entrent ensemble dans les étapes 2 et 3 du présent protocole. Modification des procédures pour C. elegans sont décrites aux étapes 4 et 5 et P. affinis d’édition est couvert dans les étapes 6 et 7. Enfin, puisque le succès d’une expérience d’édition de gène dans un organisme puisse être évalué par séquençage de génotype, sous-étapes décrivant les méthodes d’analyse possible pour toutes les cellules et les organismes décrites dans le protocole sont présentés à l’étape 8.
Protocol
1. RNP Assemblée
-
Concevoir l’expérience bien à l’avance, l’acquisition de toutes les composantes ARN, ADN et protéines avance. Comme un premier passage, essayez un des contrôles positifs énumérés au tableau 1 et utiliser des réactifs commerciaux décrits dans le tableau des matériaux afin d’assurer un dispositif expérimental fiable et l’intégrité des matériaux. Pour d’autres conseils sur la planification d’une nouvelle expérience de modification du génome, voir études sur ce sujet12,42,43.
Remarque : Une fois assemblé tel que décrit dans les étapes ultérieures, RNP, préparé à l’avance peut-être être conservés à-80 ° C.- Après avoir choisi quel gène cible, utilisez l’une des outils en ligne gratuits pour concevoir un gRNA optimal44,45,46,47,,48. N’oubliez pas de cibler un exon si l’espoir de générer un knock-out.
Remarque : Ces outils aideront à identifier un site cible avec un voisin de S. pyogenes PAM séquence, score de haute qualité et faible hors cible. - Purifier la protéine Cas9 de S. pyogenes grâce à des méthodes publiées8, ou l’acheter auprès d’un fournisseur commercial.
- Préparer un tampon Cas9 typique pour la dilution de RNA, RNP préparation et stockage de protéine, qui contient 20 mM pH HEPES 7.5, 150 mM de KCl, 10 % de glycérol et 1 mM de TCEP. Utilisez toujours l’eau exempte de nucléase dans les tampons qui seront utilisés pour remettre en suspension ou diluer RNA pour prévenir la dégradation.
- Produire le guide RNA (tracrRNA et crRNA ou sgRNA) à travers une transcription in vitro à l’aide de méthodes publiées ou achetez-la auprès d’un acide nucléique synthèse entreprise17,21,49, 50 , 51.
- Si l’insertion d’un gène, synthétiser, ou achetez un donneur matrice d’ADN.
- Stocker les protéines et les aliquotes de RNA à-80 ° C et décongélation sur glace immédiatement avant l’emploi.
Remarque : Chaque gel-dégel abaisse légèrement l’efficacité. Des protocoles détaillés, accès libre pour Cas9 purification52 et la transcription in vitro de sgRNAs53 sont disponibles ailleurs.
- Après avoir choisi quel gène cible, utilisez l’une des outils en ligne gratuits pour concevoir un gRNA optimal44,45,46,47,,48. N’oubliez pas de cibler un exon si l’espoir de générer un knock-out.
- Si vous travaillez avec le c. elegans, passez à l’étape 1.5. Pour le protocole P. affinis , passez à l’étape 1.6. Si vous utilisez sgRNA, passez à l’étape 1.4. Passez à l’étape 1.3 pour assembler un gRNA pour l’édition de cellules primaires.
-
Assembler un gRNA en mélangeant des quantités équimolaires de tracrRNA et crRNA. Faire 100 µL de 80 actions de gRNA µM, pour environ 50 expériences de modification du génome.
- Incuber le gRNA à 37 ° C pendant 30 min et ensuite laisser refroidir lentement à température ambiante.
-
Préparation à la RNP montage HSPC et T cell : assembler un complexe RNP en mélangeant très lentement, les 1-2 x molaire quantit6 de gRNA à 200 pmol de Cas9 protéine dans un volume total de 10 µL. Ajouter Cas9 concentré à la gRNA (préalablement dilué dans le tampon de Cas9) pendant environ 30 s , faisant des cercles rapides avec la pipette, ce qui porte à la concentration finale de Cas9 à 20 µM.
- Préparer les cuvettes d’électroporation.
Remarque : Ce protocole est spécifique au système commercial dénommé dans la Table des matières, mais RNP édition peut également être réalisé avec d’autres dispositifs d’électroporation. - Ajouter 5 µL (100 pmols, lymphocytes T) ou 10 µL (200 pmol, HSPCs) du RNP à chaque cuve.
- Si l’insertion d’ADN nouveau plutôt que de faire un coup de grâce, ajouter 1 µL de 100 µM (100 pmol) oligonucléotides monocaténaires donneur ADN (ssODN)25,,du5455 pour les cuvettes ou le puits de la plaque.
- Passez à l’étape 2 pour les instructions suivantes dans l’édition de cellules primaires protocole.
- Préparer les cuvettes d’électroporation.
-
Préparation à la RNP édition de c. elegans : assembler le RNP complexe en ajoutant les réactifs suivants afin de créer un volume final de 20 µL (les concentrations finales sont notées entre parenthèses) : Cas9 (2 µM), pH HEPES 7,5 (10 µM), KCl (115 µM), crRNA (12 µM) , tracrRNA (40 µM) et les modèles de réparation si nécessaire (0,5 µM ADNsb ou jusqu'à 350 ng/µL ADNdb).
Remarque : L’efficacité d’une réparation induite par le Cas9 ORD-basé sur un modèle est proportionnelle à la concentration de la construction de réparation d’ADN double brin ; ainsi, plus la concentration du modèle réparation, le plus efficace de la réparation basé sur un modèle. Cependant, une injection de mélanges contenant plus de 350 ng/µL d’ADN double brin s’est avérée réduire la viabilité vers injectée. Ainsi, il est préférable d’utiliser, mais pas plus de 350 ng/µL d’ADN double brin dans le mélange pour maximiser l’efficacité de la réparation tout en minimisant la létalité.- Ajouter plusieurs crRNAs de cibler plusieurs loci simultanément, selon les besoins pour l’approche de la co-CRISPR/co-conversion préalable décrite à l’étape 5.4. Lorsque vous ajoutez plus d’un crRNA, ajouter séquentiellement au master mix.
Remarque : La quantité de chaque crRNA n’a pas besoin d’être les mêmes, et même doublement de la concentration totale de crRNAs en master mix sans modifier la concentration de Cas9 ne semble pas interférer avec la fréquence de la mutagénèse à un lieu spécifique. Exemples sont décrits en détail dans la Paix et al. 56. - Mélanger en pipettant également et faites tourner la solution RNP à 16 000 x g pendant 5 s pour s’assurer que la solution est recueillie en bas du tube.
- Incuber la solution à 37 ° C pendant 15 m.
- Centrifuger l’échantillon à 16 000 x g pendant 1 min granuler des particules qui peuvent boucher l’aiguille mince-bored microinjection. Utiliser le liquide surnageant dans les étapes ultérieures.
- Passez à l’étape 4 pour le reste du protocole c. elegans .
- Ajouter plusieurs crRNAs de cibler plusieurs loci simultanément, selon les besoins pour l’approche de la co-CRISPR/co-conversion préalable décrite à l’étape 5.4. Lorsque vous ajoutez plus d’un crRNA, ajouter séquentiellement au master mix.
-
Préparation à la RNP affinis P. édition : préparer des aliquotes de Cas9 jetables avec eau exempte de nucléase rouge de phénol (pour la visualisation des injections) leur dilution à une concentration finale de 6,25 µM de rouge de phénol Cas9 et 0,15 %.
- Assembler le RNP complexe en mélangeant un 2-5 x excès molaire de gRNA à la protéine de Cas9 dans un volume total de 6 µl. Ajouter 12 pmol de Cas9 à gRNA, ce qui porte la concentration finale de Cas9 à 2 µM, gRNA concentration de 4 à 8 µM et la concentration de rouge de phénol à 0,05 %.
- Incuber le mélange à température ambiante pendant 10 min au complexe le RNP.
- Passez à l’étape 6 pour les instructions suivantes dans l’édition de P. affinis protocole.
2. Culture et préparation des cellules
Remarque : Effectuez des opérations 2.1.1 à 3.3.3 sous une hotte de sécurité biologique.
-
L’homme cryoconservé achat mobilisé circulants CD34+ HSPCs auprès d’un fournisseur.
- Décongeler les ~ 1 x106 HSPCs dans une eau à 37 ° C pendant 3 min de bain et transférez-les sur un tube conique de 15 mL. Ajouter 10 mL d’un milieu sans sérum expansion provenant d’une source commerciale et tourner le mélange à x 100 g pour 10 min. Retirez le surnageant et remettre en suspension les cellules dans 2 mL de SFEM supplémenté. Les cellules en plaques 6 puits de la plaque et leur culture dans un incubateur à 37 ° C pendant 24 à 48 h avant l’électroporation RNP.
- Compter les cellules avec un hémocytomètre et transfert le nombre total de HSPCs nécessaire (150 000-200 000 HSPCs par cuvette d’être électroporation) dans un tube à centrifuger.
- Tourner le tube à 100 x g pendant 10 min granuler les cellules.
-
Acheter les CD4 primaires humaines+ T cells d’un vendeur ou isoler de sang total humain par la centrifugation en gradient de densité29.
- Avant l’activation des cellules T, les enduire 48 puits culture plaques de αCD3 (UCHT1) et αCD28 (CD28.2). Enduire les plaques avec 500 µL de 10 µg/mL αCD3 et αCD28 de 10 µg/mL dans du PBS pendant au moins 2 h à 37 ° C.
Remarque : Pour certains loci, NHEJ peut être réalisée sans stimulation préalable, mais y compris cette étape maximise son efficacité. - Cellules de culture le T pendant 48 h à 37 ° C sur les plaques lié aux anticorps αCD3/αCD28 dans un milieu complet de RPMI [RPMI-1640 additionné de 5 mM de HEPES, 2 mM d’alternative commerciale à la L-Glutamine, 50 µg/mL de pénicilline/streptomycine, 50 µM de 2-mercaptoéthanol, 5 mM de les acides aminés essentiels, 5 mM de pyruvate de sodium et 10 % (vol/vol) FBS]. Cellules de culture le T à une densité de 2 000 000 cellules de T dans 500 µL de médias / puits d’une plaque de 48 puits.
- Cellules de comte le T en utilisant un hémocytomètre et transfert le nombre total de lymphocytes T nécessaires pour l’électroporation expérimenter (100 000-1 000 000 cellules de T par cuvette d’être électroporation) dans un tube à centrifuger.
- Tourner le tube à 90 x g pendant 8 min granuler les cellules. Si les cellules ont été densité gradient séparée en 2 jours, elles tournent à 200 x g pendant 8 min.
- Avant l’activation des cellules T, les enduire 48 puits culture plaques de αCD3 (UCHT1) et αCD28 (CD28.2). Enduire les plaques avec 500 µL de 10 µg/mL αCD3 et αCD28 de 10 µg/mL dans du PBS pendant au moins 2 h à 37 ° C.
-
Pour les deux types de cellules, aspirer le surnageant avec un pipette/aspirateur, enlever toutes les bulles.
- Resuspendre doucement les cellules avec 20 µL de tampon d’électroporation par cuvette.
- Ajouter 20 µL des cellules (HSPCs 150 000-200 000 ou 100 000-1 000 000 cellules de T) pour chaque cuve, qui contient déjà 10 µL de la RNP et mélanger bien en pipettant également monter et descendre sans créer de bulles.
3. RNP électroporation
- Electroporate les cuves après en les plaçant dans un nucleofector. Pour les HSPCs, utilisez le code d’impulsion ER100. Pour les cellules T, utilisez le code d’impulsion EH-115.
-
HSPCs uniquement : Ajouter 100 µL d’un milieu supplémenté de SFEM (chauffé à 37 ° C) pour chaque cuve immédiatement après l’électroporation et laisser que les cellules récupérer pendant 10-15 min.
- Les cellules de transfert à la culture dans un fond rond 96 puits sur plaque et ajouter une supplémentaire 100 µL de milieu supplémenté SFEM pendant 24 h.
- Modifiez-les à un milieu frais de SFEM complété et les incuber pendant 24 à 72 h supplémentaires.
- Supprimer les cellules pour le génotypage eux 48-96 h post-électroporation. Faites tourner les cellules à 300 g pendant 5 min et enlever le surnageant avant de commencer l’extraction de l’ADN (étape 8.2).
-
Cellules T seulement : ajouter 80 µL de RPMI toutes les milieux de culture préchauffée à 37 ° C dans le réservoir à chaque cuve ou bien, à l’aide d’une pipette multicanaux (si nécessaire).
- Les incuber à 37 ° C pendant 15 min.
- Ajouter des médias appropriés, anticorps, cytokines, etc. à la plaque d’immatriculation destination et préchauffer dans un incubateur à 37 ° C.
- Transférer 107 µL des cellules électroporation des puits d’une plaque à 96 puits fond rond à l’aide d’une pipette multicanaux (si nécessaire).
- Pour plus d’informations sur l’évaluation des résultats édition, passez à l’étape 8.
4. préparation de c. elegans
-
1 jour avant microinjection : préparer les coussinets de gel d’agarose pour la microinjection.
- Faire une solution d’agarose à 3 % (p/v) dans de l’eau en ajoutant agarose à l’eau et porter la solution à ébullition sur une plaque chauffante ou dans un four à micro-ondes.
- Disposer de 24 x 50 mm x 1,5 diapositives lamelle couvre-objet sur une table et utiliser une pipette Pasteur en verre pour mettre une goutte de petit (~ 15 µL) de solution d’agarose dans la diapositive. Aplatir rapidement la chute d’agarose en plaçant une autre lamelle couvre-objet sur le dessus. Permettre l’agarose solidifier et puis retirez une des lamelles.
- Laissez la lamelle d’agarose-enduit face vers le haut sur une table pendant la nuit pour sécher. Après 24h, stocker les coussinets de gel d’agarose dans un récipient propre et sec.
Remarque : Il peuvent être utilisés indéfiniment.
- Tirer les aiguilles de microinjection : à l’aide de capillaires en verre borosilicate avec des filaments (diamètre de 1,0 mm et intérieur diamètre extérieur 0,58 mm), tirer les aiguilles issus de Mello et feu57 et autres ressources58. Les aiguilles peuvent être utilisés immédiatement ou peuvent être stockés dans un récipient propre et sec, contreventé par des supports argile.
- Pour l’entretien vers, préparer une gélose de nématode croissance Media (NGM) versé dans les plats de Pétri et parsemées de bactéries OP50 (pour les protocoles sur le standard c. elegans entretien et recettes pour des milieux de culture, voir Stiernagle,59).
- Les vers de microinjection de scène : 12 à 24 heures avant la microinjection, choisissez hermaphrodites L4-mise en scène d’une nouvelle plaque de NG-agar avec OP50 bactéries et les incuber pendant la nuit à 20 ° C. Pour chaque mélange de cible/injection Cas9, choisissez ~ 30 vers la plaque.
-
Jour de microinjection : Charger l’aiguille tirées de microinjection avec la solution de RNP surnageante préparée à l’étape 1.5.
- Pipeter le surnageant de l’étape 1.5.4 dans une pipette capillaire extraite et le remblai, la solution de la pipette capillaire à l’aiguille de microinjection préparés (généralement moins de 0.1 µL de chargement).
- Monter l’aiguille chargé sur l’appareil de microinjection attaché à un micromanipulateur. Régler la pression d’appareil injection 250 kPa et la pression d’équilibre à 25 kPa.
-
Pause-retour la pointe de l’aiguille chargée de générer une arête de l’aiguille. Placez un 15 mm x 15 mm x 1,5 mm carré lamelle sur le dessus d’un 24 mm x 50 mm x 1,5 mm lamelle couvre-objet.
- Superposer une extrémité de la lamelle carrée avec Huilehalocarbone 700.
- Placez l’aiguille dans l’huile, au bord de la lamelle carré 15 mm.
- À l’aide d’un coup de main pour guider la platine du microscope et la lamelle, brossez la diapositive vers le haut et le long du bord de l’aiguille tout en appuyant sur la touche/pédale d’injection. Casser le dos, la pointe de l’aiguille en augmentant le flux du liquide hors de l’aiguille. Atteindre un débit optimal en faisant l’injection intervertissez écoulement le long du bord de l’aiguille, formant 1 ~ bulle/s.
- Confirmer que les vers L4 pris 12-24h avant microinjection sont développemental mise en scène de jeunes adultes le jour de l’injection. Choisissez les vers adultes jeunes pour un plat d’agar-NG qui n’a pas de bactéries OP50 et permettez-leur de ramper pendant 5 min. Cela réduit la quantité de bactéries transféré au pad injection, minimisant les sabots de l’aiguille.
- Placez une injection d’agarose pad/lamelle couvre-objet sur une étendue de la dissection. À l’aide d’une pique de ver, posez une petite piste de Huilehalocarbone sur un bord du coussin.
-
Avec le choix de ver enduit à l’huile, soulever plusieurs vers la plaque de NG-agar et sur la piste de l’huile. Avec un cheveu attaché à une pipette, par exemple un cil ou chat Moustache, positionner les vers en parallèle, doucement en poussant les vers dans le coussinet de gel d’agarose. Jusqu'à ce qu’à l’aise avec la procédure de microinjection, seulement monter et injecter un ver à la fois.
Remarque : L’agarose sec sera absorbé l’humidité vers, obligeant à adhérer au pad. Par conséquent, il faut travailler rapidement car les vers peuvent dessécher.- Une fois en position et attaché au pad, superposer les vers avec un autre prendre quelques gouttes d’huile d’halocarbures (~ 20 µL) de l’extrémité du ver.
5. c. elegans gonades Microinjection avec RNP et soin après l’injection
Remarque : Le protocole de microinjection est adapté de Mello et feu57et décrit en détail ailleurs60,61.
-
Placer la lamelle avec les vers montée sur le microscope de l’injection. Sous un faible grossissement (objectif de 5 X, 10 X oculaire), positionner les vers perpendiculaires à l’aiguille d’injection.
- Commutateur à un fort grossissement (objectif X 40, 10 X oculaire), repositionner l’aiguille adjacente au bras des gonades correspondant à la zone près des noyaux en milieu - à-stade pachytène tardif.
- En utilisant le micromanipulateur, déplacer l’aiguille contre le ver, en appuyant légèrement sur la cuticule. Puis, avec d’une part, sur le côté de la platine du microscope à secouer l’aiguille à travers la cuticule. Appuyer sur la touche/pédale injection lentement remplir le bras de gonades avec le mélange de l’injection et retirer l’aiguille.
- Répétez cette étape avec l’autre branche de la gonade.
-
Une fois que les vers sont injectés, retirer le coussin de la lamelle couvre-objet / d’agarose et placez-le sous un microscope à dissection.
- À l’aide d’une pipette capillaire extraite, remplacer l’huile vers en pipettant également un tampon M9 sur eux. Effectuer ce traitement pour libérer les vers de la gélose.
- Après 10 min, quand les vers sont volée autour dans la mémoire tampon, déplacez-les vers un NG-gélose avec OP50 bactéries à l’aide de la pipette capillaire extraite. Placer la plaque à 20 ° C pendant 2-3 h jusqu'à ce que les vers ont récupéré et sont déplacent autour.
- Une fois rétabli, individuellement, transférer les vers à NG-gélose avec OP50 et transférer les plaques dans une étuve à 25 ° C.
-
Permettre la P0-injecté vers à croître et à jeter la progéniture pendant 3 jours. La progéniture de1 F de l’écran.
- Si vous utilisez co-CRISPR ou conversion co62,63,64,65, puis sélectionnez les vers de candidat pour la sélection fondée sur le savoir si ils ont le phénotype mutant du gène référence. Individuellement transférer ces marqué vers de nouvelles plaques de NG-agar avec OP50 et leur permettre d’établir la descendance2 F à 20 ° C.
Remarque : Le phénotype utilisé pour un dépistage de co-CRISPR ou la sélection contient une estimation précoce pour le succès de l’édition de Cas9. - Si le phénotype de co-CRISPR n’est pas présent, microinject un plasmide contrôle positif pour aider à améliorer l’efficacité de microinjection.
NOTE : par exemple, y compris un plasmide dans le mélange d’injection qui encode mCherry-tag MYO-2 permettra d’évaluer l’efficacité de l’injection. Worms avec succès une injection de pCFJ90 auront certains descendants avec pharynxes fluorescents.
- Si vous utilisez co-CRISPR ou conversion co62,63,64,65, puis sélectionnez les vers de candidat pour la sélection fondée sur le savoir si ils ont le phénotype mutant du gène référence. Individuellement transférer ces marqué vers de nouvelles plaques de NG-agar avec OP50 et leur permettre d’établir la descendance2 F à 20 ° C.
- Examiner les vers de1 F de la présence des modifications souhaitées. Choisir la mère de1 F à un puits individuel d’une plaque 96 puits, lyse lui, puis examinez son ADN par amplification PCR spécifiques à insérer, analyse de séquences d’ADN ou arpenteur nucléase assay (CEL-1)66.
Remarque : Ces analyses peuvent être effectuées en utilisant un dépistage co-CRISPR/co-conversion ou autre ou la sélection régimes65,66,,du6768. - Pour plus d’informations sur l’évaluation des résultats édition, passez à l’étape 8.
6. préparation de P. affinis
- 1 jour avant la microinjection, enrichir pour les embryons au début en mettant en place un « réservoir de paire » la veille ; les femmes nouvellement séparées contiendra embryons fraîchement fécondés. Voir Rehm et al. 69 pour plus de détails.
- Le jour de la microinjection, recueillir les embryons Parhyale unicellulaires (0-4 h après fécondation) par anesthésier les femelles gravides avec 0,02 % huile de girofle dans l’eau de mer et en raclant doucement les embryons hors de sa poche incubatrice ventrale en utilisant une flamme-tiré et pipette en verre arrondie et une terne paire de pinces #3.
7. P. affinis embryon Microinjection avec RNP et soin après l’injection
- Remblayer un tube capillaire extraite avec environ 1 µL du mélange injection RNP décrit ci-dessus.
-
Permet d’azote comprimé microinject chaque embryon comme décrit dans Rehm et al. 69.
- Injecter les embryons de Parhyale sous un microscope à dissection à l’aide d’un microinjector et un micromanipulateur. Charger 1,5 µL du mélange injection dans le dos d’un tube capillaire extraite (4 pouces - 1,0 mm avec des filaments, tiré à l’aide d’une micropipette tirant appareils) à l’aide d’une pointe de pipette de microloader.
- Mettre en place l’aiguille sur les appareils d’injection et briser la pointe de l’aiguille (une très petite quantité) à l’aide d’une paire de pinces sous le coup de dissection. Calibrer le volume livré en injectant dans Huilehalocarbone 700 et en mesurant le diamètre de la bulle.
- Couper un « creux » de l’agent de durcissement à l’aide d’une lame de rasoir. Remplissez-le à moitié d’eau de mer stérilisée par filtration et alignez les embryons de Parhyale dans la fosse pour stabiliser.
- Injecter les embryons en utilisant la configuration de la microinjection, stabilisation de chaque embryon avec une paire de forceps lors de l’injection. Après l’injection, utilisez une pipette en verre pour transférer les embryons vers une boîte de Petri frais 60 mm à demi remplie d’eau de mer stérilisée par filtration.
-
Si la première division a déjà eu lieu pour former un embryon de 2 cellules (fécondation après 4 à 6 h), générer entièrement mutant animaux en injectant les deux blastomères. Pour garantir un clivage total du stade 2 cellules, co injecter les blastomères FITC ou TRITC dextran et observer que le signal est limité à un seul blastomère sous un fluorescent dissection étendue après l’injection.
- Vous pouvez également générer « demi-mutant » animaux par injection d’un seul des deux blastomères au stade 2 cellules (gauche-droite à peu près divisée selon le tissu et la position le long de l’axe A-P).
- Injecter une cellule dans un embryon de 8 cellules (post-fécondation 7,5-9 h) pour limiter l’édition à une seule couche de germe. Voir Gerberding et al. 70 pour une carte de lignées de blastomère précoce.
-
Incuber les embryons dans des récipients de culture de 60 mm (pas plus de 25 par plat), à demi remplies d’eau de mer stérilisée par filtration, « pré oxygénés » à l’aide d’un barboteur aquarium ou en agitant vigoureusement.
- Placer les plats d’embryons dans un ustensiles en plastique plus ou moins étanche doublé avec un essuie-tout humide pour conserver l’humidité et les placer dans un incubateur à 26 ° C avec un cycle lumière-obscurité de 12 h.
- Transférez les embryons survivants pour l’eau de mer vaisselle tous les jours.
Remarque : Les embryons peuvent être cultivés à température ambiante, bien qu’ils développeront beaucoup plus lentement.
-
Disséquer et fixer les embryons à différentes étapes d’une analyse de l’expression par in situ hybridation ou anticorps coloration (voir Browne et al. 71 pour un guide de mise en scène et des références supplémentaires pour la dissection et fixation72, in situ hybridation73et anticorps coloration74).
- Faire des aiguilles de dissection en enfilant un morceau plié de fil de tungstène environ 0,5 en longueur à l’extrémité de l’aiguille de l’insuline. Aiguiser l’aiguille en hydroxyde de sodium sous un courant. Utiliser une seringue de 1 mL dans la poignée de l’aiguille de la dissection.
- Remplir une cupule d’une boîte de verre de 3 puits à mi-chemin d’une solution fraîchement faites de 9 pièces PEM tampon (0,1 M pH tuyaux 6,95, 2 mM de l’EGTA, 1 mM de MgSO4), 1 élément 10 x PBS et 1 partie 32 % PFA. Placez les embryons de 3-5 dans le plat et enfoncez un petit trou dans chaque embryon, en utilisant une aiguille de tungstène forte vers poke accompagné d’une légèrement ternie à se stabiliser, permettant le jaune s’écoule et le fixateur pour courir à.
- À l’aide d’une paire d’aiguilles de tungstène affûté, doucement taquiner à l’extérieurs deux membranes entourant l’embryon Parhyale . Les disséquer dans le fixateur pour rendre les embryons plus fiable mais travail rapidement pour éviter que la membrane se fixe à l’embryon, ce qui complique le déplacement de la membrane. Laissez les embryons à la difficulté pour un total de 15-20 min pour la coloration des anticorps ou 40-50 min pour l’hybridation in situ .
- Image de nouveau-nés vivants et les analyser pour les phénotypes morphologiques et comportementales ou difficulté et leur tache pour des analyses plus détaillées. Soulever les nouveau-nés à la maturité sexuelle à 2-3 mois pour établir la knockout et lignées transgéniques (voir Kontarakis et Pavlopoulos75 pour soins de nouveau-nés et d’autres informations utiles).
8. évaluation des résultats d’édition
- Le cas échéant, recherchez un phénotype visuel ou fonctionnel dans la modification de cellules ou d’organismes.
Remarque : Ce processus varie considérablement par l’application, et quelques exemples sont décrits à la fin de leurs étapes de protocole correspondant ci-dessus. Après avoir corrigé la mutation de la drépanocytose dans HSPCs, analyser la production d’hémoglobine par les érythroblastes différenciés par CLHP (Figure 1A). Un knock-out du gène du récepteur de IL-2 dans les cellules T peut être confirmé par la coloration de surface et cytométrie en flux (Figure 1B). Afin d’évaluer les phénotypes c. elegans et P. affinis , observer la morphologie animale et le comportement sous un microscope lumineux ou fluorescent (Figures 1 et D 1). - Pour déterminer l’efficacité et le type des génomiques modifications générées, lyser les pools de cellules édités et extraire leur ADN génomique à l’aide d’une exploitation commerciale kit21.
-
Pour une estimation rapide de formation indel, PCR-amplifier au moins 200 paires de bases autour de la Coupe du site et effectuer une endonuclease1 T7 (T7E1)76 ou arpenteur (CEL-1 nucléase) dosage77.
- Si une formation indel au site Cas9 coupe ou HDR réussie sera créer ou supprimer un site de restriction connu, envisagez d’utiliser une enzyme de restriction digestion pour estimer l’édition efficacité6. Le dosage de polymorphisme de longueur de fragment restriction peut être un moyen pratique de vérifier l’efficacité si elle est disponible.
- Pour une quantification précise de l’efficacité de l’édition et la détermination des résultats édition prédominants, envoyer l’amplicon PCR pour un standard Sanger sequencing avec amorces et inverses.
Remarque : Si l’analyse d’un seul clone ou organisme, l’analyse des résultats de Sanger est simple, comme illustré dans la Figure 2A. Si l’analyse d’un pool de cellules, puis analyser les chromatogrammes avec l’outil en ligne78, comme illustré à la Figure 2B. - Pour une quantification complet et les séquences de l’édition des résultats, effectuer le séquençage en profondeur27,54, comme illustré à la Figure 2C.
- Pour évaluer un ensemble particulier de modifications hors cible, PCR-amplifier les sites hors cible prévues et les envoyer pour NGS. Pour activer la détection des translocations chromosomiques, effectuer des GUIDE-seq79 ou haut-débit, translocation de génome séquençage (HTGTS)80. Pour obtenir une image complète de l’hors-cible des montages dans une population clonale, effectuer le séquençage du génome entier (WGS)81,,du8283.
Remarque : Il existe une variété de méthodes pour quantifier les modifications du génome de sur - et hors-cible, a expliqué davantage à l’examen de divers articles84,85,86.
Representative Results
Ces expériences spectacle comment pré-assemblés Cas9 RNP peut être utilisé pour manipuler les génomes de cellules primaires et des organismes entiers. Chercheurs purifient ou achètent sgRNA et protéine Cas9, combinent les deux composantes pour former avant le complexe et introduisent le RNP dans leurs cellules ou d’organismes d’intérêt. Après avoir laissé suffisamment de temps pour l’édition pour se produire et pour la progéniture de la prochaine génération d’être né (le cas échéant), recherchez les phénotypes et/ou prélever des cellules pour le génotypage. Phénotypes peuvent être observés par les tests fonctionnels, tests d’expression, visualisation (par œil ou au microscope) ou autres méthodes, en fonction de l’expérience.
Par exemple, HSPCs qui ont été omises afin de corriger la mutation de la β-globine qui provoque la drépanocytose peuvent être différenciés en érythrocytes et testés pour la production de sain ou faucille hémoglobine27,87 (Figure 1 A). Lymphocytes T sous la direction d’assommer le gène récepteur de haute affinité IL-2, CD25 (IL2RA), peuvent être analysés par la coloration de surface et écoulement cytometry88et fonctionnellement analysés afin de détecter une signalisation réponse à la stimulation IL-2 (Figure 1B ). Les lymphocytes T peuvent aussi être reprogrammées à bien des égards cliniquement importants nécessitant une évaluation des différents phénotypes, y compris l’efficacité de HIV infection89 et les cellules de l’efficacité antitumorale in vivo de voiture-T11.
En utilisant une approche de la co-CRISPR/co-conversion préalable, vers c. elegans sont édités simultanément à deux loci62. HDR à la gène de référence dpy-10 à l’aide d’un ssODN de réparation entraîne une mutation de gain de fonction dominante facilement-incisé dpy-10 modèle. Hétérozygotes F1dpy-10(gof) animaux sont à rouleaux (Rol) et animaux homozygotes dpy-10(gof) est boulotte (Dpy). La présence du phénotype indique que Cas9 édition a eu lieu chez ces animaux et améliore les chances d’identifier un événement de modification du deuxième locus chez les animaux de1 Rol ou Dpy F. Une expérience d’édition réussie devrait se traduire par 33 à 50 % des injecté P0 vers ce qui donne 20 ou plusieurs descendants de1 F qui sont Rol ou Dpy90. Il est alors possible de choisir des animaux non-Rol de retourner dpy-10 au type sauvage et sélectionnez pour le homozygote edit d’intérêt. En règle générale, la concentration de le crRNA ciblant le gène de référence co-CRISPR devrait être la moitié que de la crRNA ciblant le gène d’intérêt. Si une modification du gène d’intérêt n’est pas valorisée, les ratios du RNAs CRISPR deux peuvent être ajustés pour augmenter les chances de récupérer la mutation souhaitée. Par exemple, augmenter la quantité de crRNA pour le gène d’intérêt par rapport à la crRNA de gènes de référence augmentera le pourcentage de worms possédant des montages dans le gène d’intérêt au sein de la population de vers qui possèdent également des modifications au locus du gène de référence. Fréquences de conversion co varient, mais les tarifs sont généralement de 20 à 60 %, produisant souvent des montages homozygotes dans la génération de1 F (Figure 1C).
P. affinis petites tortues qui ont été omises afin d’assommer le gène Abdominale-B (Abd-B) affichent les anomalies morphologiques clair3 (Figure 1D). Ce gène est nécessaire pour la bonne structuration abdominale, et ses résultats de perturbation thoracique de type saut et pattes remplaçant les jambes de natation et d’ancrage qui sont généralement présent sur l’abdomen.
Déterminer l’édition des résultats au niveau génotypique de génome nécessite le séquençage ou un test in vitro qui détecte les changements de la séquence. Nous démontrons ici les données du séquençage représentatif de nos types de cellules de modèle et d’organismes, mettant en évidence les différentes approches de l’édition de quantification. Notez que les étiquettes de la figure sont généralisés car toutes les méthodes indiquées ici peuvent être appliquées à n’importe quel système biologique.
Approches axées sur le séquençage varient en complexité technique et la profondeur des résultats. Pour des populations clonales éditées ou organismes individuels facilement séparables, individus édités peuvent être séquencés après extraction de l’ADN génomique. Résultats de séquençage Sanger standard révèlera la modification de la séquence sur le site de Cas9 coupe chez un individu donné, avec des déphasages hypothétiques qui perturberaient sa fonction (Figure 2A). L’outil en ligne pour le séquençage est une autre approche basée sur le séquençage Sanger qui peut être appliquée à une population mixte plutôt que des mutants individuels78. Séquences sont analysés avec un outil en ligne qui peut rapprocher l’efficacité globale du montage ainsi que les résultats de la séquence principale. Les données représentatives sont indiquées dans la Figure 2B.
La méthode de séquençage plus complet décrite ici est le séquençage en profondeur (parfois dénommé séquençage haut-débit ou génération). Cette méthode fournit des séquences d’ADN des génomes individuels dans une population mixte. Ces données peuvent être illustrées par une multitude de moyens. Ici, nous avons classé les lectures de séquençage individuel de cellules édités basés sur les résultats de montage (Figure 2C). La plupart des cellules sont édités par la voie NHEJ, qui se traduit généralement par perturbation de gène. Dans d’autres, le gène cible a été échangé pour une version alternative via HDR27.
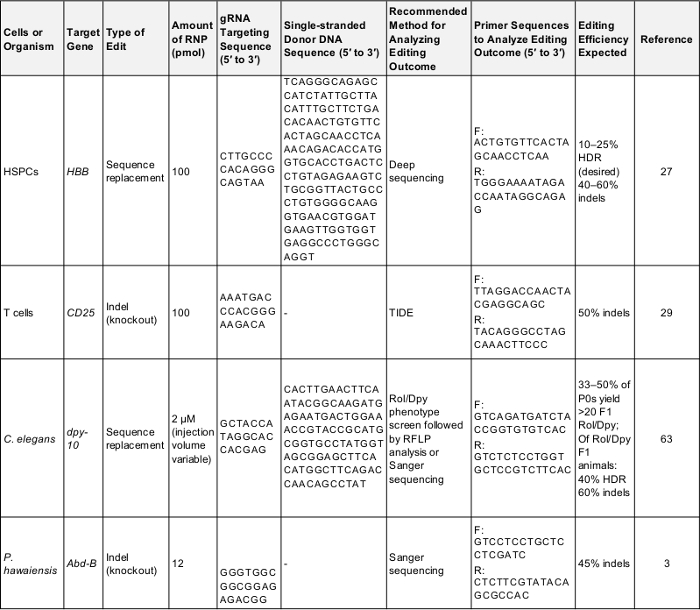
Tableau 1 : positif contrôle préliminaire génome édition expériences. Ce tableau présente les informations clées nécessaires à l’exercice d’un génome de première édition expérience dans chacune des cellules et des organismes décrites dans le présent protocole. Suivant ces paramètres est susceptible de fournir un résultat positif qui peut être utilisé pour tester le protocole ou comme point de référence pour la comparaison, une fois l’expérimentateur est ciblant un gène de leur propre intérêt. F: avant, R: inverse, HDR : réparation axés sur l’homologie. S’il vous plaît cliquez ici pour télécharger ce tableau.

Figure 1 : Représentant phénotypique résulte de Cas9 RNP éditer des cellules humaines primaires et organismes. (A) il s’agit d’une trace HPLC montrant qu’après succès génome édition, HSPCs qui sont différenciées en stades érythroblastes produira hémoglobine plus fonctionnelle que l’hémoglobine de faucille. Érythrocytes mutants produisent la faucille l’hémoglobine (HbS), tandis que-édité avec succès des cellules vont produire de l’hémoglobine sain (HbA et HbA2) ainsi que de l’hémoglobine fœtale (HbF). L’absorbance est représenté graphiquement en unités arbitraires (UA). Ce panneau a été publié en DeWitt et al. 27. il est réimprimé avec la permission de l’Association américaine pour l’avancement de la Science. (B) sur la gauche, pour chaque État, ce panneau montre les données de cytométrie en flux montrant que colorées à la surface des cellules T n’expriment pas CD25 après que le gène CD25 a été assommé avec RNP. L’abondance de CD25 est tracée sur l’axe des abscisses avec la taille de la cellule sur l’axe y. Sur la droite, pour chaque État, ce panneau montre la quantification de la Phospho-Stat5 (pStat5) après une induction avec IL-2. La signalisation est réduite lorsque le récepteur IL-2 est absente (CD25 KO). L’abondance pStat5 est tracée sur l’axe des abscisses et compare les données résultant de trois différents niveaux d’entrée IL-2 verticalement. (C) ce panneau montre un écran Caenorhabditis elegans co-CRISPR/co-conversion de ciblage dpy-10 comme le marqueur de conversion Co. Deux guide ARN cible deux loci, de dpy-10 et de votre gène favori (Kuaytiaw), dans le même P0-animal injecté. HDR à dpy-10 entraîne un phénotype Rol ou Dpy. La sélection des animaux1 Rol-f ou Dpy augmente les chances d’identifier les modifications au second locus. (D) ce panneau montre que le type sauvage Parhyale affinis nouveau-nés ont abdomen normal avec des jambes de natation et d’ancrage. Les nouveau-nés de knock out Abd-B (individus de0 F) développent un abdomen transformé vers le thorax. Ainsi, la natation et les ancres jambes sont disparus et remplacés par des pieds de saut d’obstacles et marche associées à un thorax normal. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.

Figure 2 : Résultats typiques de modifier les méthodes d’analyse des résultats. (A) ce tableau montre des exemples de la Sanger résultats du séquençage des organismes individuels de F1 P. affinis , y compris la séquence de type sauvage et trois indels différents qui perturbent la fonction du gène en déplaçant la trame de lecture ouverte. (B) marée ces résultats montrent l’éventail des insertions et des délétions qui ont eu lieu sur un site de Cas9-cible à un pool de cellules de T séquencées. L’axe x indiquent la longueur d’une donnée d’insertion ou de suppression en nucléotides. (C) ces résultats de séquençage en profondeur ne montrent aucune modification du génome sans nucleofection ou gRNA, et succès édition avec intact Cas9 RNP, regroupés par résultat de réparation de l’ADN dans HSPCs. s’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
Discussion
Établissant un génome robuste protocole d’édition dans une cellule, ligne ou organisme d’intérêt nécessite l’optimisation et empirique stable de plusieurs paramètres clés, présentées dans cette section. Essayer quelques variantes des approches générales présentées ici est fortement encouragée. La clé de la limitation de ce protocole est que l’application de ces méthodes à d’autres cellules ou organismes peuvent conduire à un résultat différent selon les espèces étudiées, et un modèle expérimental qui mène à un masquage haute efficacité gène ne peut-être pas promouvoir insertion de l’ADN. Ainsi, nous recommandons de commencer par les méthodes présentées ici et dépannage tel que décrit ci-dessous.
Dépannage du génome édition qualité réactif :
Générer ou l’achat des réactifs de haute qualité est une étape essentielle dans n’importe quel génome protocole d’édition. Cas9 protéine peut être purifiée dans le laboratoire ou acheté dans le commerce. Nombreux protocoles note une concentration finale de Cas9 dans recettes RNP, mais le gène optimal de l’activité d’édition dépendra de l’activité spécifique de toute préparation de protéines individuelle Cas9, qui varie en fonction de la source. Une fois le protocole présenté ici est au travail, examiner en optimisant la quantité de RNP utilisée par titration Cas9 niveaux pour établir une concentration optimale : celui qui fournit le clivage de l’ADN cible hautement spécifique sans clivage de hors-cible inutile causé par excessive de Cas940.
Guide RNA pureté et homogénéité peuvent également être déterminants du génome édition succès22. SgRNAs achetés ou composantes distinctes de crRNA et tracrRNA sont des réactifs de qualité en général et une variété de modifications chimiques sont disponibles pour lutter contre les problèmes de dégradation de l’ARN ou imprégner des fonctionnalités supplémentaires à la RNP91. GRNAs chimiquement modifiés n’est peut-être pas nécessaire pour génome standard édition expériences, certains groupes ont observé beaucoup plus montage efficacité avec ces réactifs, donc ils peuvent être intéressant d’essayer après avoir maîtrisé le processus et/ou lorsque la dégradation gRNA semble être une question22,91. In vitro de transcription et gel subséquent purification est une alternative peu coûteuse, qui peut être suffisante pour routine génome édition expériences17,21,49,50. En outre, plusieurs approches qui sont généralement appliquées pour produire gRNA homogène des populations in vivo, y compris ribozyme et ARNt dotés d’excision des guides individuels, peut être prolongée à in vitro préparation d’ARN pour générer le nettoyeur produits92.
Guide RNA et donateurs ADN design conseils :
Guide RNA est un facteur critique dans la réalisation d’un montage sur la cible hautement efficace tout en minimisant les chances de clivage hors cible. Pour vous aider dans la sélection du guide, plusieurs études ont utilisé des écrans de haut débit couplés avec le séquençage de prochaine génération pour compiler les caractéristiques de la séquence des guides réussi47,79,,du9394, 95,96. Ces caractéristiques ont été utilisés pour développer des algorithmes prédictifs et des outils en ligne pour aider à guide sélection44,45,46,47,48. Ces algorithmes reposent sur les écrans à l’aide de systèmes basés sur l’ADN pour guide expression RNA. Guides sont exprimées à l’aide d’un promoteur Pol III, et leur expression est donc sujette à des limitations associées à la transcription Pol III, tels que l’interruption prématurée lorsqu’elle rencontre des voies ferrées de l’uracile97,98, 99. Toutefois, le fait usage du RNP avec in vitro-guide synthétisée RNAs contourne ces préoccupations et simplifie les contraintes sur la conception du guide. Une caractéristique commune qui ont émergé de ces algorithmes et a été confirmée par de nombreuses études avec l’édition de génome très efficace, est la présence d’une purine, particulièrement une guanine, à l’extrémité 3′ de séquence spécifique à la cible du guide. Cette fonction de guide a été très réussie entre les organismes allant de mammifères à c. elegans, drosophile et zebrafish65,100,101. En outre, pour c. elegans, conception des guides avec un dinucléotide GG à l’extrémité 3′ de la région de ciblage du guide est une stratégie efficace pour prédire le guide très efficace RNAs65. Idéalement, tester plusieurs guides en parallèle afin de déterminer qui est le plus réussi pour une application donnée.
Lorsque vous tentez d’insérer une séquence d’ADN dans le génome, la conception du donneur / ADN est également cruciale. Donateurs d’oligonucléotides monocaténaires (ssODNs) sont insérés plus fiable que les autres modèles de réparation typique, linéaire double brin et plasmide ADN54,55,102. À certains locus, efficacité HDR peut être améliorée avec les ssODNs qui sont complémentaires à la non ciblées ou déplacés de brin d’ADN et possèdent des armes d’homologie qui sont asymétriques en longueur27,,55. Étant donné que le modèle de la réparation est inséré sur le site de coupe et comprend la séquence ciblée, doivent prendre des mesures pour empêcher le clivage de l’ADN du donneur avant ou après l’insertion génomique Cas9. Ceci est accompli en faisant des mutations silencieuses à la séquence de PAM ou de la région de semences, en évitant la reconnaissance par Cas9 tout en conservant la fonction du gène inséré21,103. Si les modifications de nucléotides même à la PAM sont susceptibles de supprimer la liaison104, essayer de changer au moins quatre nucléotides pour plus de sécurité.
Importance et applications futures :
Génome de montage avec CRISPR-Cas9 est devenue une méthode puissante qui permet une manipulation génétique de tout organisme. Montage avec le RNP Cas9 prend un peu plus d’effort dans un premier temps mais est facile à utiliser une fois réactifs et protocoles établis dans un laboratoire. Montage des cellules avec RNP pré-assemblés au lieu de l’ADN plasmidique mène à rendement édition global élevé, y compris l’insertion de gène difficile à atteindre par l’intermédiaire de HDR, avec effets hors cible moins24,25,26 , 27 , 29. en outre, expérimentateurs éviter des problèmes avec l’expression des gènes, RNA la dégradation, le repliement des protéines et l’association entre gRNA et Cas9 molécules synthétisées séparément au sein de la cellule22,23. RNP édition contourne également les problèmes de sécurité sur la mutagénèse insertionnelle et expression durable qui peut-être survenir lorsque les méthodes de livraison viral sont utilisés en clinique14. En raison de ces avantages, beaucoup de scientifiques menant précliniques, validation des expériences favorisent RNP d’édition pour des applications thérapeutiques humaines. In vivo et ex vivo axée sur le RNP génome édition approches sont en développement pour traiter et guérir une variété de conditions, d’une maladie génétique comme la dystrophie musculaire de Duchenne105 et drépanocytose27 au Le VIH cancer et29 11. Fait intéressant, Cas9 RNP est plus en plus utilisée comme une méthode de livraison pour l’ingénierie agricole, car elle permet de « Sans ADN » édition de plantes33,34,36.
Disclosures
Les auteurs Alexander Marson et Jacob E. Corn sont les co-fondateurs de Spotlight Therapeutics. Jacob E. Corn est un conseiller à la Mission thérapeutique et son laboratoire bénéficie du soutien du Fonds de recherche d’AstraZeneca et Pfizer. Alexander Marson est conseiller auprès de Juno Therapeutics et Pacte thérapeutique, et son laboratoire bénéficie du soutien du Fonds de recherche de Juno Therapeutics, Epinomics et Sanofi. Son laboratoire a aussi appliqué aux brevets liés à la technologie de Cas9 RNP.
Acknowledgments
Nous remercions les nombreux membres antérieures de nos laboratoires et de la communauté d’édition de génome Bay Area pour leur contribution au développement de ces méthodes. Nous remercions Ross Wilson pour la lecture critique de ce manuscrit.
Recherche de Alexander Marson est pris en charge par un don de la Jake Aronov et un National Multiple Sclerosis Society accorde (CA 1074-A-21). Alexander Marson est titulaire d’une bourse de carrière pour les chercheurs médicaux du fonds Burroughs Wellcome et Chan Zuckerberg Biohub chercheur. Recherche de Jacob E. Corn est pris en charge par le Li Ka Shing Foundation, le Heritage Medical Research Institute Medical et le California Institute for Regenerative Medicine. Behnom Farboud et de Barbara J. Meyer la recherche est financée en partie par la subvention NIGM R01 GM030702 à Barbara J. Meyer, qui est un enquêteur du Howard Hughes Medical Institute. Recherche Erin Jarvis et Nipam H. Patel est financée en partie par la subvention de NSF IOS-1257379 et Erin Jarvis reconnaît le soutien d’un GRFP NSF et une bourse d’études supérieures Philomathia.
Materials
| Name | Company | Catalog Number | Comments |
| Reagents/Materials | |||
| DNA oligonucleotides | Integrated DNA Technologies | - | IDT will provide custom DNA sequences, including those in Table 1 |
| Guide RNAs | Synthego | - | Synthego will provide high-quality sgRNAs for S. pyogenes Cas9, including custom sgRNAs containing the targeting sequences included in Table 1 |
| Purified Cas9 protein (EnGen Cas9 NLS, S. pyogenes) | New England Biosciences | M0646T | If possible, purifying Cas9 in-house or purchasing from local core facilities is a less expensive option |
| Normal peripheral blood CD34+ stem/progenitor cells | AllCells | PB032-2 | |
| StemSpan SFEM | StemCell Technologies | 09650 | |
| StemSpan CC110 | StemCell Technologies | 02697 | |
| P3 Primary Cell 4D-Nucleofector X Kit | Lonza | V4XP-3032 | |
| RPMI-1640 Medium, With sodium bicarbonate, without L-glutamine, liquid | Sigma | R0883-6X500ML | |
| EasySep™ Human T Cell Isolation Kit | Stemcell | 17951 | |
| cell culture plate, 96 wells, round | Fisher Scientific | 3799 | |
| CTS (Cell Therapy Systems) Dynabeads CD3/CD28 | Life Tech | 40203D | |
| Reombinant Human IL-2 | UCSF Pharmacy | NA | |
| SepMate-50 500-pack IVD | Stemcell Technologies | 85460 | |
| OP50 Escherichia coli | Caenorhabditis Genetics Center | OP-50 | https://cgc.umn.edu/ |
| Nematode Growth Media agar in petri dishes | - | - | See Stiernagle, T (ref. 59) |
| Standard borosilicate glass capillaries with filament: 4 in (100 mm), 1/0.58 OD/ID | World Precision Instruments | 1B100F-4 | |
| Single-barrel standard borosilicate glass capillaries: 6 in (152 mm), 2/1.12 OD/ID | World Precision Instruments | 1B200-6 | |
| Cover glass; 24 × 50 mm | Thermo Fisher Scientific | 12-544E | |
| Cover glass; 22 × 22 mm | Thermo Fisher Scientific | 12-518-105K | |
| Apex LE agarose | Genesee Scientific | 20-102 | |
| Halocarbon oil 700 | Sigma-Aldrich | H8898-100ML | |
| pCFJ90 plasmid | Addgene | 19327 | |
| Compressed nitrogen | - | ||
| 60 mM culture dishes | BD | ||
| Capillary tubes with filament: 4 in (1.0 mm) | World Precision Instruments | T2100F-4 | |
| Sylgard 184 | Dow Corning | ||
| Petri dishes (100 × 15 mm) | - | ||
| Tungsten wire (0.005 in. diameter) | Ted Pella | ||
| Perfluoroalkoxy alkane (PFA) | - | ||
| Marine salt | - | ||
| 9" pasteur pipettes | - | ||
| Phenol red | - | ||
| Nuclease-free water | - | ||
| Equipment | |||
| 4D Nucleofector | Lonza | AAF-1002X | |
| MZ75 Stereomicroscope | Leica | Out-of-production. Current model is the M80 Stereomicroscope | |
| Axio Vert35 inverted phase contrast fluorescent microscope | Zeiss | Out-of-production. Current model is the Axio VertA.1 | |
| Laser-based micropipette puller (for C. elegans protocol) | Sutter Instrument | FG-P2000 | |
| Picoliter Microinjector (for C. elegans protocol) | Warner Instruments | PLI-100A | |
| Three-axis Joystick oil hydraulic micromanipulator | Narishige International | MO-202U | |
| Coarse manipulator | Narishige International | MMN-1 | |
| Micropipette puller (for P. hawaiensis protocol) | Sutter Instrument | P-80/PC | |
| Microinjector (for P. hawaiensis protocol) | Narishige | IM300 | |
| Microloader pipette tips | Eppendorf | 5242956003 | |
| NG-agar |
References
- Komor, A. C., Badran, A. H., Liu, D. R. CRISPR-based technologies for the manipulation of eukaryotic genomes. Cell. , 1-17 (2016).
- Barrangou, R., Horvath, P. A decade of discovery: CRISPR functions and applications. Nature Microbiology. 2, 1-9 (2017).
- Martin, A., Serano, J. M., et al. CRISPR/Cas9 mutagenesis reveals versatile roles of Hox genes in crustacean limb specification and evolution. Current Biology. 26 (1), 14-26 (2016).
- Goldstein, B., King, N. The future of cell biology: emerging model organisms. Trends in Cell Biology. 26 (11), 818-824 (2016).
- Mali, P., Yang, L., et al. RNA-guided human genome engineering via Cas9. Science. 339 (6121), 823-826 (2013).
- Cong, L., Ran, F. A., et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 339 (6121), 819-823 (2013).
- Deltcheva, E., Chylinski, K., et al. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature. 471 (7340), 602-607 (2011).
- Jinek, M., Chylinski, K., Fonfara, I., Hauer, M., Doudna, J. A., Charpentier, E. A Programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 337 (6096), 816-821 (2012).
- Nowak, C. M., Lawson, S., Zerez, M., Bleris, L. Guide RNA engineering for versatile Cas9 functionality. Nucleic Acids Research. 44 (20), 9555-9564 (2016).
- Jiang, W., Cox, D., Zhang, F., Bikard, D., Marraffini, L. A. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nature Biotechnology. , 1-9 (2013).
- Rupp, L. J., Schumann, K., et al. CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Scientific Reports. 7 (1), 737 (2017).
- Graham, D. B., Root, D. E. Resources for the design of CRISPR gene editing experiments. Genome Biology. 16, 260 (2015).
- Wang, H., La Russa, M., Qi, L. S. CRISPR/Cas9 in genome editing and beyond. Annual Review of Biochemistry. 85, 2270 (2016).
- Nelson, C. E., Gersbach, C. A. Engineering delivery vehicles for genome editing. Annual Review of Chemical and Biomolecular Engineering. 7, 637-662 (2016).
- Yin, H., Kauffman, K. J., Anderson, D. G. Delivery technologies for genome editing. Nature Reviews Drug Discovery. 16 (6), 387-399 (2017).
- Zuris, J. A., Thompson, D. B., et al. Cationic lipid-mediated delivery of proteins enables efficient protein-based genome editing in vitro and in vivo. Nature Biotechnology. 33 (1), 73-80 (2015).
- Cho, S. W., Lee, J., Carroll, D., Kim, J. -S., Lee, J. Heritable gene knockout in Caenorhabditis elegans by direct injection of Cas9-sgRNA ribonucleoproteins. Genetics. 195 (3), 1177-1180 (2013).
- Wang, W., Kutny, P. M., et al. Delivery of Cas9 protein into mouse zygotes through a series of electroporation dramatically increases the efficiency of model creation. Journal of Genetics and Genomics. 43 (5), 319-327 (2016).
- Chen, S., Lee, B., Lee, A. Y. -F., Modzelewski, A. J., He, L. Highly efficient mouse genome editing by CRISPR ribonucleoprotein electroporation of zygotes. Journal of Biological Chemistry. 291 (28), 14457-14467 (2016).
- Remy, S., Chenouard, V., et al. Generation of gene-edited rats by delivery of CRISPR/Cas9 protein and donor DNA into intact zygotes using electroporation. Scientific Reports. 7 (1), 16554 (2017).
- DeWitt, M. A., Corn, J. E., Carroll, D. Genome editing via delivery of Cas9 ribonucleoprotein. Methods. , 1-7 (2017).
- Hendel, A., Bak, R. O., et al. Chemically modified guide RNAs enhance CRISPR-Cas genome editing in human primary cells. Nature Biotechnology. 33 (9), 985-989 (2015).
- Thyme, S. B., Akhmetova, L., Montague, T. G., Valen, E., Schier, A. F. Internal guide RNA interactions interfere with Cas9-mediated cleavage. Nature Communications. 7, 11750 (2016).
- Kim, S., Kim, D., Cho, S. W., Kim, J., Kim, J. -S. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Research. 24 (6), 1012-1019 (2014).
- Lin, S., Staahl, B. T., Alla, R. K., Doudna, J. A. Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. eLife. 3, 04766 (2014).
- Liang, X., Potter, J., et al. Rapid and highly efficient mammalian cell engineering via Cas9 protein transfection. Journal of Biotechnology. 208, 44-53 (2015).
- DeWitt, M. A., Magis, W., Bray, N. L., Wang, T. Selection-free genome editing of the sickle mutation in human adult hematopoietic stem/progenitor cells. Science Translational Medicine. 8 (360), (2016).
- Ramakrishna, S., Kwaku Dad, A. -B., Beloor, J., Gopalappa, R., Lee, S. -K., Kim, H. Gene disruption by cell-penetrating peptide-mediated delivery of Cas9 protein and guide RNA. Genome Research. 24 (6), 1020-1027 (2014).
- Schumann, K., Lin, S., et al. Generation of knock-in primary human T cells using Cas9 ribonucleoproteins. Proceedings of the National Academy of Sciences of the United States of America. 112 (33), 10437-10442 (2015).
- Lee, J. -S., Kwak, S. -J., et al. RNA-guided genome editing in Drosophila with the purified Cas9 protein. G3: Genes, Genomes, Genetics (Bethesda, MD). 4 (7), 1291-1295 (2014).
- Sung, Y. H., Kim, J. M., et al. Highly efficient gene knockout in mice and zebrafish with RNA-guided endonucleases. Genome Research. 24 (1), 125-131 (2014).
- Menoret, S., De Cian, A., et al. Homology-directed repair in rodent zygotes using Cas9 and TALEN engineered proteins. Scientific Reports. 5, 14410 (2015).
- Woo, J. W., Kim, J., et al. DNA-free genome editing in plants with preassembled CRISPR-Cas9 ribonucleoproteins. Nature Biotechnology. 33 (11), 1162-1164 (2015).
- Malnoy, M., Viola, R., et al. DNA-free genetically edited grapevine and apple protoplast using CRISPR/Cas9 ribonucleoproteins. Frontiers in Plant Science. 7, 1904 (2016).
- Svitashev, S., Schwartz, C., Lenderts, B., Young, J. K., Mark Cigan, A. Genome editing in maize directed by CRISPR-Cas9 ribonucleoprotein complexes. Nature Communications. 7, 13274 (2016).
- Liang, Z., Chen, K., et al. Efficient DNA-free genome editing of bread wheat using CRISPR/Cas9 ribonucleoprotein complexes. Nature Communications. 8, 14261 (2017).
- Shin, S. -E., Lim, J. -M., et al. CRISPR/Cas9-induced knockout and knock-in mutations in Chlamydomonas reinhardtii. Scientific Reports. 6, 27810 (2016).
- Pohl, C., Kiel, J. A. K. W., Driessen, A. J. M., Bovenberg, R. A. L., Nygård, Y. CRISPR/Cas9 based genome editing of Penicillium chrysogenum. ACS Synthetic Biology. 5 (7), 754-764 (2016).
- Grahl, N., Demers, E. G., Crocker, A. W., Hogan, D. A. Use of RNA-protein complexes for genome editing in non-albicans Candida species. mSphere. 2 (3), (2017).
- Rivera-Torres, N., Kmiec, E. B. A standard methodology to examine on-site mutagenicity as a function of point mutation repair catalyzed by CRISPR/Cas9 and ssODN in human cells. Journal of Visualized Experiments. (126), (2017).
- Nandal, A., Mallon, B., Telugu, B. P. Efficient generation and editing of feeder-free IPSCs from human pancreatic cells using the CRISPR-Cas9 system. Journal of Visualized Experiments. (129), (2017).
- Mohr, S. E., Hu, Y., Ewen-Campen, B., Housden, B. E., Viswanatha, R., Perrimon, N. CRISPR guide RNA design for research applications. The FEBS Journal. 283 (17), 3232-3238 (2016).
- Bauer, D. E., Canver, M. C., Orkin, S. H. Generation of genomic deletions in mammalian cell lines via CRISPR/Cas9. Journal of Visualized Experiments. (95), e52118 (2015).
- Hsu, P. D., Scott, D. A., et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nature Biotechnology. 31 (9), 827-832 (2013).
- Heigwer, F., Kerr, G., Boutros, M. E-CRISP: fast CRISPR target site identification. Nature Methods. 11 (2), 122-123 (2014).
- Moreno-Mateos, M. A., Vejnar, C. E., et al. CRISPRscan: designing highly efficient sgRNAs for CRISPR-Cas9 targeting in vivo. Nature Methods. 12 (10), 982-988 (2015).
- Labun, K., Montague, T. G., Gagnon, J. A., Thyme, S. B., Valen, E. CHOPCHOP v2: a web tool for the next generation of CRISPR genome engineering. Nucleic Acids Research. 44, 272-276 (2016).
- Haeussler, M., Schönig, K., et al. Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biology. 17 (1), 148 (2016).
- Lo, T. -W., Pickle, C. S., et al. Precise and heritable genome editing in evolutionarily diverse nematodes using TALENs and CRISPR/Cas9 to engineer insertions and deletions. Genetics. 195 (2), 331-348 (2013).
- Bassett, A., Liu, J. -L. CRISPR/Cas9 mediated genome engineering in Drosophila. Methods. 69 (2), 128-136 (2014).
- Prior, H., Jawad, A. K., MacConnachie, L., Beg, A. A. Highly efficient, rapid and co-CRISPR independent genome editing in Caenorhabditis elegans. G3: Genes, Genomes, Genetics. , Bethesda, MD. (2017).
- Hirsh, A. Cas9 expression and purification protocol. protocols.io. , (2017).
- DeWitt, M. A., Wong, J. In vitro transcription of guide RNAs. protocols.io. , (2017).
- Yang, L., Guell, M., et al. Optimization of scarless human stem cell genome editing. Nucleic Acids Research. 41 (19), 9049-9061 (2013).
- Richardson, C. D., Ray, G. J., DeWitt, M. A., Curie, G. L., Corn, J. E. Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA. Nature Biotechnology. 34 (3), 339-344 (2016).
- Paix, A., Folkmann, A., Seydoux, G. Precision genome editing using CRISPR-Cas9 and linear repair templates in C. elegans. Methods. 121-122, 86-93 (2017).
- Mello, C., Fire, A. DNA transformation. Methods in Cell Biology. 48, 451-482 (1995).
- Sutter Pipette Cookbook. , Available from: https://www.sutter.com/PDFs/pipette_cookbook.pdf (2017).
- Stiernagle, T. Maintenance of C. elegans. WormBook: the online review of C. elegans biology. , (2006).
- Evans, T. C. Transformation and microinjection. WormBook: the online review of C. elegans biology. , (2006).
- Berkowitz, L. A., Knight, A. L., Caldwell, G. A., Caldwell, K. A. Generation of stable transgenic C. elegans using microinjection. Journal of Visualized Experiments. (18), (2008).
- Kim, H., Ishidate, T., et al. A co-CRISPR strategy for efficient genome editing in Caenorhabditis elegans. Genetics. 197 (4), 1069-1080 (2014).
- Arribere, J. A., Bell, R. T., Fu, B. X. H., Artiles, K. L., Hartman, P. S., Fire, A. Z. Efficient marker-free recovery of custom genetic modifications with CRISPR/Cas9 in Caenorhabditis elegans. Genetics. 198 (3), 837-846 (2014).
- Ward, J. D. Rapid and precise engineering of the Caenorhabditis elegans genome with lethal mutation co-conversion and inactivation of NHEJ repair. Genetics. 199 (2), 363-377 (2015).
- Farboud, B., Meyer, B. J. Dramatic enhancement of genome editing by CRISPR/Cas9 through improved guide RNA design. Genetics. 199 (4), 959-971 (2015).
- Wood, A. J., Lo, T. -W., et al. Targeted genome editing across species using ZFNs and TALENs. Science. 333 (6040), 307 (2011).
- Friedland, A. E., Tzur, Y. B., Esvelt, K. M., Colaiácovo, M. P., Church, G. M., Calarco, J. A. Heritable genome editing in C. elegans via a CRISPR-Cas9 system. Nature Methods. 10 (8), 741-743 (2013).
- Dickinson, D. J., Ward, J. D., Reiner, D. J., Goldstein, B. Engineering the Caenorhabditis elegans genome using Cas9-triggered homologous recombination. Nature Methods. 10 (10), 1028-1034 (2013).
- Rehm, E. J., Hannibal, R. L., Chaw, R. C., Vargas-Vila, M. A., Patel, N. H. Injection of Parhyale hawaiensis blastomeres with fluorescently labeled tracers. Cold Spring Harbor Protocols. 2009 (1), (2009).
- Gerberding, M., Browne, W. E., Patel, N. H. Cell lineage analysis of the amphipod crustacean Parhyale hawaiensis reveals an early restriction of cell fates. Development (Cambridge, England). 129 (24), 5789-5801 (2002).
- Browne, W. E., Price, A. L., Gerberding, M., Patel, N. H. Stages of embryonic development in the amphipod crustacean, Parhyale hawaiensis. Genesis. 42 (3), New York, NY. 124-149 (2005).
- Rehm, E. J., Hannibal, R. L., Chaw, R. C., Vargas-Vila, M. A., Patel, N. H. Fixation and dissection of Parhyale hawaiensis embryos. Cold Spring Harbor Protocols. 2009 (1), (2009).
- Rehm, E. J., Hannibal, R. L., Chaw, R. C., Vargas-Vila, M. A., Patel, N. H. In situ hybridization of labeled RNA probes to fixed Parhyale hawaiensis embryos. Cold Spring Harbor Protocols. 2009 (1), (2009).
- Rehm, E. J., Hannibal, R. L., Chaw, R. C., Vargas-Vila, M. A., Patel, N. H. Antibody staining of Parhyale hawaiensis embryos. Cold Spring Harbor Protocols. 2009 (1), (2009).
- Kontarakis, Z., Pavlopoulos, A. Transgenesis in non-model organisms: the case of Parhyale. Methods in Molecular Biology. 1196, Clifton, NJ. 145-181 (2014).
- Kim, H. J., Lee, H. J., Kim, H., Cho, S. W., Kim, J. -S. Targeted genome editing in human cells with zinc finger nucleases constructed via modular assembly. Genome Research. 19 (7), 1279-1288 (2009).
- Qiu, P., Shandilya, H., D'Alessio, J. M., O'Connor, K., Durocher, J., Gerard, G. F. Mutation detection using Surveyor nuclease. BioTechniques. 36 (4), 702-707 (2004).
- Brinkman, E. K., Chen, T., Amendola, M., van Steensel, B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Research. 42 (22), 168 (2014).
- Tsai, S. Q., Zheng, Z., et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nature Biotechnology. 33 (2), 187-197 (2015).
- Frock, R. L., Hu, J., Meyers, R. M., Ho, Y. -J., Kii, E., Alt, F. W. Genome-wide detection of DNA double-stranded breaks induced by engineered nucleases. Nature Biotechnology. 33 (2), 179-186 (2015).
- Smith, C., Gore, A., et al. Whole-genome sequencing analysis reveals high specificity of CRISPR/Cas9 and TALEN-based genome editing in human iPSCs. Cell Stem Cell. 15 (1), 12-13 (2014).
- Veres, A., Gosis, B. S., et al. Low incidence of off-target mutations in individual CRISPR-Cas9 and TALEN targeted human stem cell clones detected by whole-genome sequencing. Cell Stem Cell. 15 (1), 27-30 (2014).
- Kim, D., Bae, S., et al. Digenome-seq: genome-wide profiling of CRISPR-Cas9 off-target effects in human cells. Nature Methods. 12 (3), 237-243 (2015).
- Hendel, A., Fine, E. J., Bao, G., Porteus, M. H. Quantifying on- and off-target genome editing. Trends in Biotechnology. 33 (2), 132-140 (2015).
- O'Geen, H., Yu, A. S., Segal, D. J. How specific is CRISPR/Cas9 really. Current Opinion in Chemical Biology. 29, 72-78 (2015).
- Tsai, S. Q., Joung, J. K. Defining and improving the genome-wide specificities of CRISPR-Cas9 nucleases. Nature Reviews Genetics. 17 (5), 300-312 (2016).
- Hoban, M. D., Cost, G. J., et al. Correction of the sickle cell disease mutation in human hematopoietic stem/progenitor cells. Blood. 125 (17), 2597-2604 (2015).
- Simeonov, D. R., Gowen, B. G., et al. Discovery of stimulation-responsive immune enhancers with CRISPR activation. Nature. , (2017).
- Hultquist, J. F., Schumann, K., et al. A Cas9 ribonucleoprotein platform for functional genetic studies of HIV-host interactions in primary human T cells. Cell Reports. 17 (5), 1438-1452 (2016).
- Paix, A., Wang, Y., et al. Scalable and versatile genome editing using linear DNAs with microhomology to Cas9 sites in Caenorhabditis elegans. Genetics. 198 (4), 1347-1356 (2014).
- Lee, K., Mackley, V. A., et al. Synthetically modified guide RNA and donor DNA are a versatile platform for CRISPR-Cas9 engineering. eLife. 6, (2017).
- Minkenberg, B., Wheatley, M., Yang, Y. CRISPR/Cas9-enabled multiplex genome editing and its application. Progress in Molecular Biology and Translational Science. 149, 111-132 (2017).
- Doench, J. G., Fusi, N., et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nature Biotechnology. 34 (2), 184-191 (2016).
- Doench, J. G., Hartenian, E., et al. Rational design of highly active sgRNAs for CRISPR-Cas9-mediated gene inactivation. Nature Biotechnology. , 1-8 (2014).
- Liu, H., Wei, Z., Dominguez, A., Li, Y., Wang, X., Qi, L. S. CRISPR-ERA: a comprehensive design tool for CRISPR-mediated gene editing, repression and activation. Bioinformatics (Oxford, England). 31 (22), 3676-3678 (2015).
- Wu, X., Scott, D. A., et al. Genome-wide binding of the CRISPR endonuclease Cas9 in mammalian cells. Nature Biotechnology. 32 (7), 670-676 (2014).
- Bogenhagen, D. F., Brown, D. D. Nucleotide sequences in Xenopus 5S DNA required for transcription termination. Cell. 24 (1), 261-270 (1981).
- Cozzarelli, N. R., Gerrard, S. P., Schlissel, M., Brown, D. D., Bogenhagen, D. F. Purified RNA polymerase III accurately and efficiently terminates transcription of 5S RNA genes. Cell. 34 (3), 829-835 (1983).
- Chen, B., Gilbert, L. A., et al. Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell. 155 (7), 1479-1491 (2013).
- Gagnon, J. A., Valen, E., et al. Efficient mutagenesis by Cas9 protein-mediated oligonucleotide insertion and large-scale assessment of single-guide RNAs. PLoS ONE. 9 (5), 98186 (2014).
- Ren, X., Yang, Z., et al. Enhanced specificity and efficiency of the CRISPR/Cas9 system with optimized sgRNA parameters in Drosophila. Cell Reports. 9 (3), 1151-1162 (2014).
- Ran, F. A., Hsu, P. D., Wright, J., Agarwala, V., Scott, D. A., Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nature Protocols. 8 (11), 2281-2308 (2013).
- Serano, J. M., Martin, A., et al. Comprehensive analysis of Hox gene expression in the amphipod crustacean Parhyale hawaiensis. Developmental Biology. 409 (1), 297-309 (2016).
- Sternberg, S. H., Redding, S., Jinek, M., Greene, E. C., Doudna, J. A. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature. , 1-17 (2014).
- Lee, K., Conboy, M., et al. Nanoparticle delivery of Cas9 ribonucleoprotein and donor DNA in vivo induces homology-directed DNA repair. Nature Biomedical Engineering. 1 (11), 889-901 (2017).
