

Summary
Utilizando um Cas9 preassembled ribonucleoprotein complexo (RNP) é um método poderoso para edição de genoma preciso e eficiente. Aqui, destacamos a sua utilidade em uma ampla variedade de células e organismos, incluindo células humanas primárias e os dois clássicos e emergentes organismos modelo.
Abstract
Site-specific do genoma eucariótico edição com CRISPR (clusterizado regularmente intercaladas curta palíndromo se repete)-sistemas de Cas (CRISPR-associado) rapidamente se tornou um lugar-comum entre os investigadores perseguindo uma grande variedade de questões biológicas. Os usuários mais frequentemente empregam a proteína Cas9 derivada de Streptococcus pyogenes em um complexo com um guia facilmente reprogramado RNA (gRNA). Esses componentes são introduzidos em células, e através de uma base de emparelhamento com uma região complementar do genoma de DNA (dsDNA) de dupla-hélice, a enzima cliva as duas vertentes para gerar uma ruptura da dobro-Costa (DSB). Reparação subsequente leva à inserção aleatória ou eventos de exclusão (puntuais) ou a incorporação de DNA experimentador fornecido no local da ruptura.
O uso de um único-guia do RNA e Cas9 proteína purified, preassembled para formar um RNP e entregues directamente às células, é uma potente abordagem para alcançar a edição de gene altamente eficiente. Edição de RNP particularmente aumenta a taxa de inserção do gene, um resultado que muitas vezes é um desafio para alcançar. Em comparação com a entrega de um plasmídeo, a persistência mais curta da RNP Cas9 dentro da célula conduz a menos eventos fora do alvo.
Apesar de suas vantagens, muitos usuários casuais de CRISPR gene edição estão menos familiarizados com esta técnica. Para diminuir a barreira de entrada, nós esboçamos protocolos detalhados para a implementação da estratégia da RNP em uma variedade de contextos, destacando seus benefícios distintos e diversas aplicações. Cobrimos a edição em dois tipos de células humanas primárias, células T e células tronco/progenitoras hematopoiéticas (HSPCs). Também mostramos como RNP Cas9 edição permite a fácil manipulação genética de organismos inteiras, incluindo a lombriga modelo clássico Caenorhabditis elegans e o mais recentemente introduzido crustáceo modelo, Parhyale hawaiensis.
Introduction
Fo sistema CRISPR-Cas9 permite aos cientistas alter regiões alvo de qualquer genoma1. Esta tecnologia rápida e barata tem revolucionado a pesquisa básica e promete tornar-se um profundo impacto sobre o desenvolvimento de terapias personalizadas doença, agricultura de precisão e para além de2. CRISPR edição é uma ferramenta de democratização e implementação do sistema em um novo laboratório não requer nenhuma perícia particular, no genoma engenharia básica biologia molecular habilidades. Os investigadores agora podem estudar organismos anteriormente intratáveis com alguns meios alternativos para manipulação genética3,4. Nos últimos cinco anos sozinhos, CRISPR genoma edição serviu para engenheiro de mais de 200 diferentes vertebrados, invertebrados, plantas e espécies microbianas.
Adaptado a partir da via de defesa procarióticas CRISPR, os elementos essenciais necessários para site-specific genoma edição são a proteína Cas9, normalmente a partir de S. pyogenes e códon otimizado com um sinal de localização nuclear adicionado (NLS) e seus especializados Guia de RNA5,6. Embora não discutidos aqui, outros Cas9 orthologues ou endonucleases CRISPR também podem ser utilizadas. A gRNA natural é composto de duas partes separadamente transcritos, o RNA CRISPR (crRNA) e o trans-ativando crRNA (tracrRNA)7. Estes RNAs podem ser fundidos em um único transcrito, conhecido como o single-guia do RNA (sgRNA)8. A maioria dos editores de genoma escolher o simplificada sgRNA9, embora o dual-guia também é usado regularmente10,11. Experimentadores escolher um alvo de DNA genômico 20-nucleotide (nt), garantindo que fica ao lado de uma assinatura de licenciamento curta necessária para reconhecimento de Cas9, chamado um motivo adjacente protospacer (PAM) e projetar uma gRNA que contém a sequência complementar12 .
Uma vez dentro da célula, a RNP complexo localiza seu alvo genômico, os pares de base de gRNA com o DNA complementar da costa, e então a enzima cliva as duas cadeias de ADN para gerar um dobro-Costa quebrar2. Maquinaria de reparo celular corrige o DSB, um de pelo menos duas rotas: via não-homóloga propenso fim-adesão (NHEJ) via ou o reparo de homologia-dirigido (HDR), que incorpora perfeitamente o DNA contendo 'armas' da homologia para ambos os lados da ruptura. O caminho de reparação ex normalmente leva a indel formação e rompimento de gene consequente, enquanto este último permite experimentadores inserir ou alterar as sequências de DNA1.
A edição de eficiência e precisão dependem dos meios pelo qual Cas9 e gRNA entrar dentro da célula. Esses componentes podem ser entregues para culturas de células, embriões ou organismos sob a forma de ácidos nucleicos ou como um preassembled RNP complexo13,14,15. Métodos de entrega baseada em ácido nucleico comuns incluem a transdução viral, transfeccao ou electroporation do mRNA ou plasmídeo. Guia de RNA e proteína Cas9 então são produzidos dentro da célula e se associam para formar um complexo.
A entrega direta da RNP requer a purificação separada da proteína Cas9 e guia do RNA. Isto pode ser feito em casa, ou a proteína e o sgRNA podem ser comprados de um dos vários fornecedores comerciais. Uma vez adquirido, o Cas9 e gRNA são misturados para formar o complexo RNP enzimaticamente competente e apresentados a células por injeção direta em embriões e ovos fertilizados, baseada em lipídios transfeccao16ou eletroporação. O primeiro relatório da RNP edição injeção envolvida no c. elegans gônadas17. Microinjeção ainda é o meio preferido de introduzir RNP em embriões e organismos de todo, embora electroporation eficaz tem sido demonstrado em embriões de20 do rato18,19 e rato. Descrevemos os protocolos para injetar diretamente RNP em c. elegans gônadas e p. hawaiensis embriões e recomendar um tipo especializado de eletroporação para entregar RNP ao editar células humanas primárias. Este método, nucleofection, envolve programas electroporation otimizado e soluções específicas do tipo de célula e permite a RNP entrar tanto no citoplasma e o núcleo21.
Edição de genoma com RNP oferece várias vantagens distintas. Porque os componentes de RNA e proteína são pré-montados e qualidade pode ser assegurada antes da entrega, RNP edição evita muitas armadilhas associadas a entrega à base de ácido nucleico. Ou seja, não há risco de integração Cas9-codificação do DNA no genoma do hospedeiro, mRNA nunca é exposto para degradação, e contorna problemas na vivo gRNA ou proteína expressão, dobramento e Associação22,23. Além disso, usando RNP leva à baixa toxicidade e muito menos eventos fora do alvo do que a expressão baseados no plasmídeo, um resultado do Half-Life mais curto da RNP dentro da célula24,25,26,27.
Finalmente, comprovadamente RNP edição leva a altas taxas de edição em uma variedade de linhas de células humanas, as células primárias tais como fibroblastos, células-tronco embrionárias (CES), induzida por células-tronco pluripotentes (iSPCs), HSPCs, e T células16,24, 25,26,,27,28,29; em invertebrados, incluindo c. elegans, hawaiensis p.e as moscas de fruta3,17,30; em espécies de vertebrados como zebrafish, ratos e ratos31,32; em plantar espécies incluindo Arabidopsis, tabaco, alface, arroz, videira, maçã, milho e trigo33,34,35,36; e em Chlamydomonas, Penicilliume espécies de Candida 37,38,39. A frequência de formação indel pode ser maior quando se utiliza a RNP em comparação com a entrega do plasmídeo, e inserção de DNA mediada por HDR pode ser mais fácil de conseguir25,,27,29.
O protocolo descrito aqui usa o RNP Cas9 e é uma técnica eficaz, facilmente adaptável que é simples de aplicar a uma ampla variedade de40,sistemas biológicos41, especialmente nas células que são difíceis de trabalhar com e em organismos sem sistemas bem estabelecidos para manipulação genética exacta. Começamos por descrever como criar, obter e montar a RNP Cas9 antes cobrindo seu uso através de organismos e tipos de células diferentes do modelo. Tronco/progenitoras hematopoiéticas células (HSPCs) e células T são editadas usando o mesmo método, nucleofection, então eles são cobertos juntos nas etapas 2 e 3 do presente protocolo. Procedimentos de edição para C. elegans são descritos nas etapas 4 e 5 e P. hawaiensis edição é coberto nas etapas 6 e 7. Finalmente, uma vez que o sucesso de uma experiência de edição de gene em qualquer organismo pode ser avaliado por sequenciamento de genótipo, sub-etapas descrevendo métodos de análise possível para todas as células e organismos descritos no protocolo são descritas na etapa 8.
Protocol
1. a RNP Assembly
-
Projete o experimento com grande antecedência, adquirindo todos os componentes do RNA, DNA e proteína antes do tempo. Como um passe de primeiro, tente um dos controles positivos listados na tabela 1 e usar os reagentes comerciais descritos na tabela de materiais para garantir um projeto experimental de confiança e a integridade dos materiais. Dicas adicionais sobre planejamento de uma nova experiência de edição de genoma, ver artigos sobre este tópico12,42,43.
Nota: Uma vez montado conforme descrito nas etapas subsequentes, RNPs preparados antecipadamente, podem ser armazenados a-80 ° C.- Depois de escolher qual gene alvo, use uma das ferramentas on-line gratuito para projetar um ideal gRNA44,,45,46,47,48. Certifique-se de direcionar um exon se na esperança de gerar um nocaute.
Nota: Estas ferramentas ajudará a identificar um local de destino com um adjacente S. pyogenes PAM sequência, pontuação de alta qualidade e baixa pontuação fora do alvo. - Purificar a S. pyogenes Cas9 proteínas através de de métodos publicados8, ou comprá-lo de um fornecedor comercial.
- Prepare um típico Cas9 buffer para diluição de RNA, RNP preparação e armazenamento de proteínas, que contém 20 mM do pH HEPES 7.5, 150mm de KCl 10% de glicerol e 1 mM de TCEP. Não utilize água livre de nuclease buffers que serão usados para Ressuspender ou diluir o RNA para prevenir a degradação.
- Produzir o guia do RNA (tracrRNA e crRNA ou sgRNA), através de uma transcrição em vitro usando métodos publicados, ou compre-a de um ácido nucleico síntese empresa17,21,49, 50 , 51.
- Se inserir um gene, sintetizar ou compra um doador modelo de DNA.
- Armazene a proteína e alíquotas de RNA a-80 ° C e degelo no gelo imediatamente antes do uso.
Nota: Cada congelamento-descongelamento ligeiramente reduz a eficiência. Detalhadas, acesso aberto protocolos para Cas9 purificação52 e a transcrição em vitro de sgRNAs53 estão disponíveis em outro lugar.
- Depois de escolher qual gene alvo, use uma das ferramentas on-line gratuito para projetar um ideal gRNA44,,45,46,47,48. Certifique-se de direcionar um exon se na esperança de gerar um nocaute.
- Se trabalhando com c. elegans, pule para a etapa 1.5. Para o protocolo de p. hawaiensis , pule para a etapa de 1.6. Se usando sgRNA, pule para a etapa 1.4. Prossiga para a etapa 1.3 para montar uma gRNA para edição de célula primária.
-
Monte uma gRNA misturando uma quantidade equimolar de tracrRNA e crRNA. Fazer 100 µ l de 80 estoque de gRNA µM, para cerca de 50 experimentos de edição do genoma.
- Incubar a gRNA a 37 ° C por 30 min e depois deixe-o arrefecer lentamente à temperatura.
-
Preparem a RNP HSPC e T célula edição: montar um complexo de RNP misturando um 1-2 x quantidade molar de gRNA para 200 pmol de proteína Cas9 em um volume total de 10 µ l... muito lentamente, adicionar Cas9 concentrado para a gRNA (pré-diluído no buffer Cas9) por cerca de 30 s , fazendo círculos rápidos com a pipeta, trazendo a concentração final de Cas9 a 20 µM.
- Prepare as cubetas de eletroporação.
Nota: Este protocolo é específico para o sistema comercial conhecido da Tabela de materiais, mas RNP edição também pode ser conseguido com outros dispositivos de eletroporação. - Adicione 5 µ l (100 pmoles, células T) ou 10 µ l (200 pmol, HSPCs) da RNP para cada cubeta.
- Se inserir o novo DNA ao invés de fazer um nocaute, adicionar 1 µ l de 100 µM (100 pmol) doador do oligonucleotide single-stranded DNA (ssODN)25,54,55 de cubetas ou cavidades da placa.
- Passe para a etapa 2 para as instruções na próxima edição da célula primária protocolo.
- Prepare as cubetas de eletroporação.
-
Preparação da RNP para edição de c. elegans : montar a RNP complexo adicionando os seguintes reagentes a fim de criar um volume final de 20 µ l (as concentrações finais são anotadas entre parênteses): Cas9 (2 µM), pH HEPES 7,5 (10 µM), KCl (115 µM), crRNA (12 µM) , tracrRNA (40 µM) e o reparo modelos se necessário (0,5 µM ssDNA ou até 350 dsDNA ng / µ l).
Nota: A eficiência de uma reparação mediada por Cas9 DSB-modelo é proporcional à concentração de construção reparação de dsDNA; assim, quanto maior a concentração do modelo de reparação, o mais eficiente a reparação com modelo. No entanto, uma injeção de misturas contendo superior a 350 ng / µ l de dsDNA tem demonstrada reduzir a viabilidade dos vermes injetados. Assim, é melhor usar, mas não mais de 350 ng / µ l de dsDNA no mix de maximizar a eficiência de reparação, minimizando sua letalidade.- Adicione vários crRNAs para direcionar loci múltiplos simultaneamente, conforme necessário para a abordagem de co-CRISPR/co-conversion triagem descrita no passo 5.4. Ao adicionar mais de um crRNA, adicione cada um sequencialmente para a mistura de mestre.
Nota: A quantidade de cada crRNA não precisa ser o mesmo, e até mesmo dobrando a concentração total de crRNAs no mix mestre sem alterar a concentração de Cas9 parece não interferir com a frequência de mutagênese em um locus específico. Exemplos são descritos detalhadamente na Paix et al 56. - Misture por pipetagem e girar a solução RNP a 16.000 x g, durante 5 s para garantir que a solução é recolhida na parte inferior do tubo.
- Incube a solução a 37 ° C por 15 m.
- Centrifugar a amostra a 16.000 x g por 1 min de partículas que podem entupir a agulha fina-furado microinjeção de Pelotas. Use o sobrenadante nas etapas subsequentes.
- Pule para o passo 4 para o restante do protocolo c. elegans .
- Adicione vários crRNAs para direcionar loci múltiplos simultaneamente, conforme necessário para a abordagem de co-CRISPR/co-conversion triagem descrita no passo 5.4. Ao adicionar mais de um crRNA, adicione cada um sequencialmente para a mistura de mestre.
-
Preparem a RNP hawaiensis p. edição: preparar alíquotas de uso único Cas9 diluindo com água livre de nuclease e vermelho de fenol (para a visualização de injeções) a uma concentração final de 6,25 µM de vermelho de fenol Cas9 e 0,15%.
- Monte a RNP complexo misturando um 2-5 x molar excesso de gRNA à proteína Cas9 em um volume total de 6 µ l. Adicionar 12 pmol de Cas9 a gRNA, trazendo a concentração final de Cas9 de 2 µM, gRNA concentração de 4-8 µM e concentração de vermelho de fenol a 0,05%.
- Incube a mistura à temperatura ambiente durante 10 min ao complexo da RNP.
- Passe para a etapa 6 para as próximo instruções na edição hawaiensis p. protocolo.
2. cultura e preparação da pilha
Nota: Execute etapas 2.1.1 para 3.3.3 em uma câmara de segurança biológica.
-
Criopreservado humanos compra mobilizaram sangue periférico CD34 + HSPCs de um fornecedor.
- Descongelar o ~ 1 x106 HSPCs em água a 37 ° C por 3 min de banho e transferi-los para um tubo cônico de 15 mL. Adicionar 10 mL de meio livre de soro de expansão de uma fonte comercial e girar a mistura a 100 x g por 10 min. Retire o sobrenadante e ressuspender as células em 2 mL de SFEM completada. As células em placas de 6-poços da placa e cultura-los numa incubadora 37 ° C por 24-48 h antes da RNP eletroporação.
- Contar as células com um hemocytometer e transfira o número total de HSPCs necessário (150.000-200.000 HSPCs por cuvete para ser electroporated) para um tubo de centrífuga.
- Gire o tubo a 100 x g durante 10 minutos para as células de Pelotas.
-
Comprar humana CD4 primário+ T células de um fornecedor ou isolá-los do sangue total humano pela centrifugação gradiente de densidade29.
- Antes da ativação de células T, pré-revestir as placas de cultura de 48-bem com αCD3 (UCHT1) e αCD28 (CD28.2). Revestir as placas com 500 µ l de 10 µ g/mL αCD3 e αCD28 de 10 µ g/mL em PBS pelo menos 2 h a 37 ° C.
Nota: Para alguns loci, NHEJ pode ser alcançada sem pre-estimulação, mas incluindo esta etapa maximiza sua eficiência. - Células de cultura o T por 48 h a 37 ° C em placas de anticorpo-limite αCD3/αCD28 em meio RPMI completa [RPMI-1640 suplementado com 5 mM de HEPES, 2mm de alternativa comercial de L-glutamina, 50 µ g/mL de penicilina/estreptomicina, 50 µM de 2-Mercaptoetanol, 5 mM de Não aminoácidos essenciais, 5mm de piruvato de sódio e 10% (vol/vol) FBS]. Células de cultura o T em uma densidade de 2.000.000 células T em 500 µ l de mídia por alvéolo de uma placa de 48.
- Contagem do T células usando um hemocytometer e transferência o número total de células T, necessários para a eletroporação experimentar (100.000-1.000.000 células T por cuvete para ser electroporated) para um tubo de centrifugação.
- Gire o tubo a 90 x g por 8 min para as células de Pelotas. Se as células foram densidade gradiente separada dentro de 2 dias, girá-los a 200 x g por 8 min.
- Antes da ativação de células T, pré-revestir as placas de cultura de 48-bem com αCD3 (UCHT1) e αCD28 (CD28.2). Revestir as placas com 500 µ l de 10 µ g/mL αCD3 e αCD28 de 10 µ g/mL em PBS pelo menos 2 h a 37 ° C.
-
Para ambos os tipos de célula, Aspire o sobrenadante com um pipeta/vácuo, remover quaisquer bolhas.
- Resuspenda suavemente as células com 20 µ l de tampão de eletroporação por cubeta.
- Adicione 20 µ l de células (150.000-200.000 HSPCs ou 100.000-1.000.000 T cells) para cada cubeta, que já contém 10 µ l da RNP e misture bem por pipetagem para cima e para baixo sem criar bolhas.
3. RNP Electroporation
- Electroporate as cubetas após colocá-los em um nucleofector. Para os HSPCs, use o código de pulso ER100. Para as células T, use o código de pulso EH-115.
-
HSPCs só: Adicione 100 µ l de uma medium SFEM completado (aquecido a 37 ° C) para cada cubeta imediatamente após electroporation e deixar que as células recuperar para 10-15 min.
- Transferi as células de cultura-los em um fundo redondo 96 poços da placa e adicionar um adicionais 100 µ l do meio SFEM complementado por 24 h.
- Mudá-los para um meio fresco de SFEM complementado e incube-os por um adicional 24-72 h.
- Retire as pilhas para a genotipagem-los 48-96 h post-eletroporação. Girar as células a 300 x g por 5 min e retire o sobrenadante antes de iniciar a extração de DNA (passo 8.2).
-
T células apenas: Adicionar 80 µ l de RPMI completa de meios de cultura, pre-aquecido a 37 ° C do reservatório a cada cubeta ou bem, usando uma pipeta multicanal (se necessário).
- -Incube a 37 ° C por 15 min.
- Adicionar a mídia adequada, anticorpos, citocinas, etc. para as placas de destino e pré-aquecê-los numa incubadora 37 ° C.
- Transferi 107 µ l das células electroporated dos poços de uma placa de 96 poços de fundo redondo usando uma pipeta multicanal (se necessário).
- Para obter mais informações sobre a avaliação dos resultados de edição, pule para a etapa 8.
4. preparação de c. elegans
-
1 dia antes da microinjeção: preparar as almofadas de agarose para o microinjection.
- Fazer uma solução de agarose de 3% (p/v) em água, adicionando agarose para água e trazendo a solução para ferver em uma chapa quente ou no microondas.
- Organizar 24 x 50 mm x 1,5 mm capa lâminas em uma tabela e usar um pipeta Pasteur de vidro para colocar uma gota de pequeno (~ 15 µ l) de solução de agarose para o slide. Rapidamente, achate a queda de agarose colocando outra lamela em cima. Permitir que a agarose solidificar e remover um das lamelas.
- Deixe a lamela de agarose-revestida virada para cima sobre uma mesa durante a noite para secar. Após 24h, armazene as almofadas de agarose em um recipiente limpo e seco.
Nota: Estes podem ser usados indefinidamente.
- Puxe as agulhas de microinjeção: usando capilares de vidro borosilicato com filamentos (diâmetro de 1,0 mm e interno de diâmetro exterior 0,58 mm), puxe as agulhas baseadas em Mello e fogo57 e outros recursos de58. As agulhas podem ser usadas imediatamente ou podem ser armazenadas em um recipiente limpo e seco, apoiado por suportes de argila.
- Para a manutenção dos vermes, preparar um ágar de nematódeo crescimento Media (NGM) vertido em placas de Petri e manchado com bactérias OP50 (para protocolos no padrão c. elegans manutenção e receitas para os meios de crescimento, ver Stiernagle,59).
- Os vermes para microinjeção de palco: 12-24 h antes da microinjeção, escolher hermafroditas L4-encenado para uma nova placa de ágar-NG com bactérias OP50 e incube-os durante a noite a 20 ° C. Para cada mistura de destino/injeção Cas9, pick ~ 30 vermes para a placa.
-
Dia da microinjeção: Carregar a agulha microinjeção puxado com a solução de RNP sobrenadante preparada no passo 1.5.
- Pipete o sobrenadante da etapa 1.5.4 em uma pipeta capilar distendida e aterre a solução da pipeta capilar para a agulha de microinjeção preparado (geralmente menos de 0,1 µ l de carga).
- Monte a agulha carregada para o aparelho de microinjeção anexado a um micromanipulador. Ajustar a pressão do aparelho de injeção para 250 kPa e a pressão de equilíbrio de 25 kPa.
-
Quebre volta a ponta da agulha carregada para gerar uma borda afiada da agulha. Coloque uma 15 x 15 mm x 1,5 mm quadrados lamela no topo de uma 24 mm x 50 mm x 1,5 mm lamela.
- Sobreposição de uma extremidade da lamela quadrada com óleo halocarbono 700.
- Posição da agulha no óleo, na borda da lamela 15 milímetros quadrados.
- Usando uma mão para guiar o microscópio e lamela, escove o slide acima e ao longo da borda da agulha enquanto pressiona o pedal de injeção/botão. Quebre a ponta da agulha, aumentando o fluxo do líquido fora a agulha. Alcançar uma taxa de fluxo ideal, tornando a injeção misturar o fluxo ao longo da borda da agulha, formando ~ 1 bolha/s.
- Confirme que os vermes de L4 escolhidos 12-24 h antes da microinjeção são adultos jovens desenvolvente encenados no dia da injeção. Escolher os vermes adultos jovens para uma placa de ágar-NG que carece de OP50 bactérias e permitir que rastejar por 5 min. Isso reduz a quantidade de bactérias transferidas para a almofada de injeção, minimizando obstruções de agulha.
- Coloque uma almofada de injeção agarose/lamela em um escopo de dissecação. Usando uma picareta de worm, colocar uma faixa pequena de óleo halocarbono ao longo de uma borda da almofada.
-
Usando a picareta de worm revestida em óleo, levante vários vermes fora a placa de ágar-NG e na faixa de óleo. Com um cabelo fino, ligado a uma pipeta, como um cílio ou gato dos whiskers, posicione os vermes em paralelo, gentilmente empurrando os vermes para a almofada de agarose. Até confortável com o procedimento de microinjeção, só montar e injetar uma minhoca de cada vez.
Nota: A agarose seco vai pavio-a umidade do vermes, levando-os a aderir à almofada. Por conseguinte, um deve trabalhar rapidamente como os vermes podem desidratar.- Uma vez em posição e fixado à almofada, sobrepor os vermes com outro escolher algumas gotas de óleo halocarbono (~ 20 µ l) da ponta do worm.
5. c. elegans Gonad Microinjection com RNPs e cuidados pós-injeção
Nota: O protocolo de microinjeção é adaptado de Mello e fogo57e descrito em detalhes em outro lugar de60,61.
-
Coloca a lamela com os vermes montados sobre o microscópio de injeção. Em um pequeno aumento (5 X objectivo ocular 10x), posicione os vermes perpendiculares a agulha de injeção.
- Interruptor para uma alta ampliação (40 X objectivo ocular 10x), reposicionar a agulha adjacente para o braço de gônada correspondente à região perto dos núcleos em meados - a tarde-paquíteno.
- Usando o micromanipulador, mova a agulha contra o worm, deprimente a cutícula ligeiramente. Em seguida, com uma mão, toque o lado do palco microscópio para abalar a agulha através da cutícula. Pressione o pedal de injeção/botão e lentamente enche o braço de gônada com a mistura de injeção e retire a agulha.
- Repita essa etapa com o outro braço da gônada.
-
Uma vez que os vermes são injetados, retirar o forro da lamela/agarose e colocá-la sob um microscópio de dissecação.
- Usando uma pipeta capilar puxada, deslocar o óleo do vermes pipetando um buffer M9 sobre eles. Realize este tratamento para liberar os vermes de agar.
- Depois de 10 min, quando os vermes são espernear no buffer, movê-los para uma placa de ágar-NG com bactérias OP50 usando a pipeta capilar puxada. Coloque a placa a 20 ° C, durante 2-3h até os vermes se recuperaram e estão se movendo ao redor.
- Uma vez recuperado, individualmente, transferir os vermes para placas de ágar-NG com OP50 e transferir as placas para uma incubadora de 25 ° C.
-
Permitir que o P0-injetado vermes para crescer e não dar descendência por 3 dias. Tela a prole de1 F.
- Se usando a conversão de co ou co-CRISPR62,63,64,,65, em seguida, selecione os vermes de candidato para a seleção com base em terem o fenótipo mutante do gene da referência. Individualmente, transferir esses vermes marcados para novas placas de ágar-NG com OP50 e permitir que leigos progênie de F2 a 20 ° C.
Nota: O fenótipo usado para um co-CRISPR seleção ou seleção deve fornecer uma estimativa inicial para o sucesso da edição de Cas9. - Se o fenótipo co-CRISPR não estiver presente, microinjeção de um plasmídeo controlo positivo para ajudar a melhorar a eficiência de microinjeção.
Nota: por exemplo, incluindo um plasmídeo no mix injeção que codifica MYO-2 mCherry-tag ajudará a avaliar a eficiência de injeção. Worms injetado com sucesso com pCFJ90 terá uma prole com pharynxes fluorescentes.
- Se usando a conversão de co ou co-CRISPR62,63,64,,65, em seguida, selecione os vermes de candidato para a seleção com base em terem o fenótipo mutante do gene da referência. Individualmente, transferir esses vermes marcados para novas placas de ágar-NG com OP50 e permitir que leigos progênie de F2 a 20 ° C.
- Examine os vermes de1 F a presença das edições desejadas. Escolher a mãe de1 F a um poço individual de uma placa de 96 poços, lyse-la e examinar o DNA por amplificação por PCR inserção específica, análise de sequências de ADN ou agrimensor nuclease ensaio (CEL-1)66.
Nota: Estes ensaios podem ser executados utilizando um co-CRISPR/co-conversion ou outra triagem ou seleção regimes65,,66,67,68. - Para obter mais informações sobre a avaliação dos resultados de edição, pule para a etapa 8.
6. preparação hawaiensis p.
- 1 dia antes a microinjeção, enriquecer para os embriões adiantados instituindo um reservatório' par' na noite anterior; as fêmeas recém separadas conterá embriões fertilizados na hora. Ver Rehm et al 69 para obter detalhes.
- No dia da microinjeção, recolher os embriões de Parhyale unicelulares (0-4 h pós fertilização) por anestesiar gravid fêmeas com óleo de cravo de 0,02% na água do mar e raspando suavemente os embriões fora de sua bolsa incubadora ventral usando uma flama-puxado e pipeta de vidro arredondado e um par sem graça de fórceps #3.
7. p. hawaiensis embrião Microinjection com RNPs e cuidados pós-injeção
- Aterramento de um tubo capilar puxado com cerca de 1 µ l do mix injeção RNP descrito acima.
-
Use nitrogênio comprimido para microinjeção cada embrião, conforme descrito em Rehm et al 69.
- Injete os embriões Parhyale sob um microscópio dissecação usando um microinjector e um micromanipulador. Carga de 1,5 µ l do mix injeção dentro de um tubo capilar puxado (4 polegadas - 1,0 mm com filamentos, puxado usando uma micropipeta puxando o aparelho) usando uma ponta de pipeta microloader.
- Configurar a agulha sobre o aparelho de injeção e quebrar a ponta da agulha (uma quantidade muito pequena), usando um par de pinças no âmbito de dissecação. Calibre o volume entregado injetando óleo halocarbono 700 e medindo o diâmetro da bolha.
- Corte uma 'calha' fora o agente de cura, usando uma lâmina de barbear. Preenchê-lo no meio do caminho com água do mar esterilizada de filtro e alinhar os embriões de Parhyale na calha para estabilizar.
- Injete os embriões usando a configuração de microinjeção, estabilizando cada embrião com um par de pinças durante a injeção. Após a injeção, use uma pipeta de transferência de vidro para transferir os embriões para um prato de cultura fresco 60 mm enchido pela metade com água do mar esterilizada de filtro.
-
Se a primeira divisão já ocorreu para formar um embrião de 2 células (pós-fertilização de 4-6 h), gere animais totalmente mutante injetando os dois blastômeros. Para garantir uma clivagem total da fase 2-célula, co injetar os blastômeros com FITC ou TRITC dextrano e observar que o sinal é restrito a um único blastômero sob uma fluorescente dissecando o escopo após a injeção.
- Como alternativa, gere 'meia-mutante' animais através da injeção de um os dois blastômeros na fase 2-célula (aproximadamente dividida esquerda-direita dependendo do tecido e da posição ao longo do eixo A-P).
- Injete uma célula em um embrião de 8 células (post-fertilização de 7,5-9 h) para restringir a edição de uma única camada de germe. Ver. Gerberding et al 70 para o mapa de linhagens de blastômero precoce.
-
Incube os embriões em pratos de cultura de 60mm (não mais que 25 por prato), preenchidos pela metade com água do mar filtro esterilizado, 'pre-oxigenado' usando um borbulhador de aquário ou agitando vigorosamente.
- Coloque os pratos de embriões em um plasticware vagamente-selado, forrada com toalha de papel molhada para manter a umidade e colocá-los numa incubadora de 26 ° C com um ciclo de claro-escuro de 12 h.
- Transferi os embriões sobreviventes para limpar pratos de água do mar todos os dias.
Nota: Embriões podem ser cultivados em temperatura ambiente, embora eles irão desenvolver muito mais lentamente.
-
Dissecar e corrigir os embriões em vários estágios para uma análise de expressão em situ da hibridação ou anticorpo coloração (veja Browne et al 71 para um guia de preparo e referências adicionais para dissecção e fixação72, em situ da hibridação73e anticorpo que mancha a74).
- Fazer agulhas de dissecação rosqueando um pedaço dobrado de fio de tungstênio aproximadamente 0.5 em de comprimento para o fim de uma agulha de insulina. Aguça a agulha sob uma corrente de hidróxido de sódio. Use uma seringa de 1 mL, como a alça da agulha de dissecação.
- Encher um poço de um prato de vidro 3-bem no meio do caminho com uma solução acabadas de 9 partes PEM tampão (0,1 M de pH tubos 6,95, 2mm de EGTA, 1 mM de MgSO4) e PBS 10 x 1-parte 1 parte 32% PFA. Coloque o prato 3-5 embriões e fazer um buraco pequeno em cada embrião, usando uma agulha afiada de tungstênio para picar e uma pouco entorpecida para estabilizar, permitindo que a gema a fluir para fora e o fixador para executar.
- Usando um par de agulhas de tungstênio afiado, delicadamente afastado arreliar as exteriores duas membranas que cercam o embrião de Parhyale . Dissecá-los em fixador para tornar os embriões mais robusto mas trabalho rapidamente para impedir que a membrana se tornar fixo para o embrião, o que torna mais difícil a remoção da membrana. Permitir que os embriões de correção para um total de 15-20 min. para coloração de anticorpo ou 40-50 min para hibridação in situ .
- Os filhotes ao vivo de imagem e analisá-los para fenótipos morfológicos e comportamentais ou corrigir e manchá-las para análises mais detalhadas. Criar os filhotes a maturidade sexual em 2-3 meses para estabelecer o nocaute e linhas transgénicas (ver Kontarakis e Pavlopoulos75 para cuidados do filhote e outros detalhes úteis).
8. avaliar resultados de edição
- Se for o caso, procure um fenótipo visual ou funcional no editado células ou organismos.
Nota: Este processo pode variar extensamente pelo aplicativo, e alguns exemplos são descritos no final de suas etapas relevantes do protocolo acima. Depois de corrigir a mutação falciforme em HSPCs, analise a produção de hemoglobina por eritroblastos diferenciados usando HPLC (Figura 1A). Um knockout do gene do receptor IL-2 em células T pode ser confirmada pela coloração da superfície e fluxo cytometry (Figura 1B). Para avaliar o c. elegans e p. hawaiensis fenótipos, observe o animal morfologia e comportamento sob um microscópio de luz ou fluorescente (figuras 1 e 1 D). - Para determinar a eficiência e o tipo das genômicas edições gerado, lisar as piscinas de células editadas e extrair seu DNA genômico usando um kit de extração comercial21.
-
Para uma estimativa rápida da indel formação, PCR-amplificação de pelo menos 200 pares de bases ao redor do corte do site e realizar um T7 endonuclease1 (T7E1)76 ou agrimensor (nuclease CEL-1) do ensaio77.
- Se uma formação indel no site Cas9-corte ou HDR bem sucedida irá criar ou remover um site de restrição conhecido, considere o uso de uma enzima de restrição digestão para estimar a eficiência edição6. O ensaio de polimorfismo (RFLP) de comprimento de fragmento de restrição pode ser uma maneira conveniente para verificar a eficiência se ele passa a ser disponível.
- Para uma quantificação exacta da edição eficiência e determinação de resultados edição predominantes, enviar o amplicon PCR para um padrão Sanger sequenciando com primers para diante e reversos.
Nota: Se analisando um único clone ou o organismo, a análise dos resultados de Sanger é simples, conforme demonstrado na Figura 2. Se analisar um pool de células, em seguida, analise os cromatogramas com a ferramenta on-line78, conforme mostrado na Figura 2B. - Para uma quantificação completa e sequências de resultados de edição, execute sequenciamento profunda27,54, conforme representado na Figura 2C.
- Para avaliar um determinado conjunto de mudanças fora do alvo, PCR-amplificar os sites fora do alvo previstos e enviá-los para NGS. Para habilitar a detecção de translocações cromossómicas, execute guia-seq79 ou elevado-throughput, translocação de todo o genoma de sequenciamento (HTGTS)80. Para um quadro completo das edições fora do alvo em uma população clonal, realize o sequenciamento do genoma inteiro (WGS)81,,82,83.
Nota: Há uma variedade de métodos para quantificar as edições do genoma na - e fora do alvo, explicadas ainda mais na revisão de vários artigos84,85,86.
Representative Results
Estas experiências, mostrar como pre-assembled Cas9 RNP pode ser usado para manipular os genomas de células primárias e organismos de todo. Pesquisadores purificam ou compram sgRNA e proteína Cas9, combinam os dois componentes para pré-forma o complexo e introduzir a RNP em suas células ou um organismo de interesse. Após permitir tempo suficiente para edição para ocorrer e para a prole da próxima geração nascer (se aplicável), procurar fenótipos e/ou recolher pilhas para genotipagem. Fenótipos podem ser observados através de ensaios funcionais, ensaios de expressão, visualização (a olho nu ou com microscopia) ou outros métodos, dependendo da experiência.
Por exemplo, HSPCs que foram editadas para corrigir a mutação de β-globina que causa a doença falciforme podem ser diferenciadas em eritrócitos e analisadas para a produção de saudável ou falciforme hemoglobina27,87 (Figura 1 A). Edição de células T para nocautear o gene do receptor de alta afinidade IL-2, CD25 (IL2RA), podem ser analisados pela coloração da superfície e fluxo cytometry88e funcionalmente analisados para detectar uma sinalização resposta à estimulação IL-2 (Figura 1B ). As células T também podem ser reprogramadas de muitas formas clinicamente importantes que exigem avaliação dos fenótipos diferentes, incluindo a eficácia do HIV infecção89 células e e na vivo eficácia antitumoral do carro-T11.
Usando uma abordagem co-CRISPR/co-conversion triagem, c. elegans vermes são editados simultaneamente em dois loci62. HDR no gene de referência dpy-10 usando um ssODN reparar os resultados do modelo em uma mutação de ganho-de-função dominante marcou facilmente dpy-10 . Heterozigotos animais dedpy-10(gof) F1são rolo (Rol) e animais homozigotos dpy-10(gof) são atarracado (Dpy). A presença do fenótipo indica que Cas9 edição ocorreu nestes animais e melhora as chances de identificar um evento de edição para o segundo locus nos animais1 Rol ou Dpy F. Uma experiência bem sucedida de edição deve resultar em 33-50% de injetado P0 vermes rendendo 20 ou mais descendentes de1 F que são Rol ou Dpy90. É possível escolher animais não-Rol retornar dpy-10 a sua e selecionar para a edição de homozigota de interesse. Como regra geral, a concentração da crRNA como alvo o gene de referência co-CRISPR deve ser meio que da crRNA como alvo o gene de interesse. Se um edit no gene de interesse não é recuperado, os rácios dos dois RNAs CRISPR podem ser ajustados para aumentar a probabilidade de recuperar a mutação desejada. Por exemplo, aumentando a quantidade de crRNA para o gene de interesse em relação a referência gene crRNA irá aumentar a percentagem de worms possuindo edições no gene de interesse dentro da população de vermes que possuem também edições no locus do gene de referência. As frequências co conversão variam, mas as taxas são tipicamente 20-60%, muitas vezes produzindo edições homozigotos na geração1 F (Figura 1C).
P. hawaiensis filhotes que foram editados para nocautear o gene Abdominal-B (Abd-B) exibir anormalidades morfológicas claro3 (Figura 1D). Este gene é necessário para a correta padronização abdominal, e seus resultados de rompimento em torácica tipo pulando e andando de pernas substituindo as pernas natação e âncora que são normalmente presentes no abdômen.
Determinação do genoma edição resultados a nível genotípico requer sequenciamento ou um ensaio em vitro que detecta alterações de sequência. Aqui, nós mostramos dados representativos de sequenciamento de nossos tipos de célula de modelo e organismos, destacando diferentes abordagens para quantificação de edição. Note-se que os rótulos da figura são generalizados porque todos os métodos mostrados aqui podem ser aplicados a qualquer sistema biológico.
Abordagens baseadas em sequenciamento variam em complexidade técnica e profundidade dos resultados. Para populações editadas clonais ou organismos individuais facilmente separável, indivíduos editados podem ser sequenciados após extração de DNA genômica. Resultados de sequenciamento Sanger padrão irão revelar a mudança de sequência no site Cas9-corte em um determinado indivíduo, com frameshifts hipotético que interromperiam sua função(Figura 2). A ferramenta on-line usada para sequenciamento é outro Sanger sequenciamento abordagem que pode ser aplicada a populações mistas, em vez de mutantes individuais78. As sequências são analisadas com uma ferramenta online que pode aproximar a eficiência geral da edição, bem como os resultados de sequência predominante. Os dados representativos são mostrados na Figura 2B.
O mais completo método de sequenciamento descrito aqui é profundo sequenciamento (por vezes referido como elevado-produção ou geração de sequenciamento). Este método fornece sequências de DNA de genomas individuais em uma população mista. Desses dados podem ser ilustrados em uma variedade de maneiras. Aqui, podemos ter classificado leituras individuais de sequenciamento de células editadas com base no resultado edição (Figura 2C). A maioria das células são editados através do caminho NHEJ, que geralmente resulta na interrupção do gene. Em outros, o gene alvo tem sido trocado por uma versão alternativa através de HDR27.
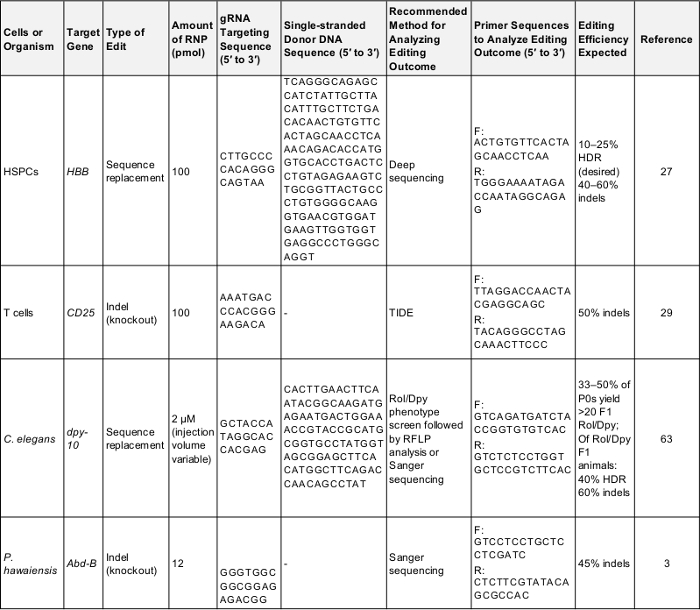
Tabela 1: positivo controla para preliminar do genoma experimentos de edição. Esta tabela mostra as principais informações necessárias para realizar um genoma pela primeira vez, edição de experiência em cada uma das células e organismos descritos no presente protocolo. Seguir estes parâmetros é susceptível de produzir um resultado bem sucedido que pode ser usado para testar o protocolo ou como base para comparação, uma vez o experimentador tem como alvo um gene de seu próprio interesse. F: encaminhar, r: reverso, HDR: reparação de homologia-dirigido. Clique aqui para baixar esta tabela.

Figura 1 : Representante fenotípica resulta da RNP Cas9 edição de células humanas primárias e organismos. (A) este é um traço HPLC mostrando que depois de genoma bem sucedida edição, HSPCs são diferenciadas em estágio final eritroblastos irá produzir hemoglobina mais funcional do que a hemoglobina de foice. Mutantes eritrócitos produzem hemoglobina falciforme (HbS), editados com sucesso células produzirá saudável hemoglobina (HbA e HbA2) assim como a hemoglobina fetal (HbF). A absorvância é graficamente em unidades arbitrárias (au). Este painel foi primeiramente publicado em DeWitt et al. 27. é reproduzido com permissão da associação americana para o avanço da ciência. (B) à esquerda, para cada condição, este painel mostra dados de citometria de fluxo mostrando que as células T de superfície manchada não express CD25 após o CD25 gene foi nocauteado com RNP. A abundância de CD25 é plotada no eixo x com o tamanho da célula no eixo y. À direita, para cada condição, este painel mostra a quantificação de fosfo-Stat5 (pStat5) após uma indução com IL-2. A sinalização é reduzida quando o receptor de IL-2 está ausente (CD25 KO). A abundância de pStat5 é plotada no eixo x e os dados resultantes de três diferentes níveis de entrada de IL-2 são comparados verticalmente. (C) este painel mostra uma Caenorhabditis elegans co-CRISPR/co-conversion tela direcionamento dpy-10 como o marcador de conversão co. Dois: guia de loci de alvo dois RNAs, dpy-10 e seu gene favorito (yfg), no mesmo P0-animal injetado. HDR em dpy-10 resulta um Rol ou Dpy fenótipo. A seleção de animais de1 Rol - ou Dpy-F aumenta as chances de identificar edições no segundo locus. (D) este painel mostra que sua Parhyale hawaiensis filhotes abdomes normais com pernas natação e âncora. Os filhotes de nocaute de Abd-B (indivíduos de0 F) desenvolvem um abdômen transformado-se no sentido do tórax. Assim, a natação e as pernas de âncoras são ido e substituídas pelas pernas pula e andando associadas com um tórax normal. Clique aqui para ver uma versão maior desta figura.

Figura 2 : Resultados típicos de edição de métodos de análise de resultado. (A) este painel mostra exemplos de Sanger a resultados de sequenciamento de F1 p. hawaiensis organismos individuais, incluindo a sua sequência e três diferentes puntuais que perturbam a função do gene deslocando o frame de leitura aberto. (B) maré estes resultados mostram a gama de inserções e eventos de exclusão que ocorreram num local Cas9-alvo em um pool de sequenciado células T. O eixo x indica o comprimento de uma determinada inserção ou exclusão de nucleotídeos. (C) estes resultados de sequenciamento profunda mostraram sem edição de genoma sem nucleofection ou gRNA e bem sucedida edição com intacto Cas9 RNP, agrupadas por resultado de reparação do DNA em HSPCs. por favor clique aqui para ver uma versão maior desta figura.
Discussion
Que institui um genoma robusto protocolo de edição em uma célula, linha ou do organismo de interesse requer a otimização e empírico testes de vários parâmetros-chave, discutidos nesta seção. Algumas variações das abordagens gerais aqui apresentadas é altamente encorajados. A limitação fundamental do presente protocolo é que aplicar esses métodos a outras células ou organismos podem levar a um resultado diferente, dependendo da espécie estudada, e um projeto experimental que leva a um nocaute do gene de alta eficiência não pode promover a inserção de DNA. Assim, recomendamos começar com os métodos aqui apresentados e solução de problemas, conforme descrito abaixo.
Solucionando problemas de genoma edição qualidade reagente:
De produção ou aquisição de reagentes de alta qualidade é uma etapa crítica em qualquer genoma protocolo de edição. Cas9 proteína pode ser purificada no laboratório ou adquirida comercialmente. Muitos protocolos nota uma concentração final de Cas9 em receitas RNP, mas o gene ideal edição atividade dependerá da atividade específica de qualquer preparação de proteína Cas9 individual, que varia dependendo da fonte. Uma vez que o protocolo apresentado aqui está funcionando, considere otimizando a quantidade de RNP usado pelos níveis de Cas9 titulados para estabelecer uma concentração ideal: aquele que fornece a clivagem de DNA alvo altamente específico sem clivagem de fora do alvo desnecessária causada por excessiva Cas940.
Guia do RNA pureza e homogeneidade também podem ser determinantes do genoma edição sucesso22. SgRNAs comprados ou componentes separados, crRNA e tracrRNA são reagentes geralmente alta qualidade e uma variedade de modificações químicas estão disponíveis para combater os problemas com a degradação do RNA ou impregnar recursos adicionais para a RNP91. Enquanto gRNAs quimicamente modificado pode não ser necessário para genoma padrão edição de experimentos, alguns grupos têm observado muito maiores eficiência com tais reagentes, de edição, assim que podem ser vale a pena tentar depois de dominar o processo e/ou quando gRNA degradação Parece ser uma questão22,91. In vitro a transcrição e subsequente gel purificação é uma alternativa barata, que pode ser suficiente para a rotina do genoma edição experiências17,21,,49,50. Além disso, várias abordagens que são comumente aplicados para produzir gRNA homogênea populações na vivo, incluindo excisão ribozima e tRNA baseada de guias individuais, pode ser estendido para in vitro preparação do RNA para gerar o aspirador produtos de92.
Guia RNA e DNA de doador de design dicas:
Seleção de RNA guia é um fator crítico na realização altamente eficiente de edição no alvo, minimizando as chances de clivagem fora do alvo. Para ajudar na seleção de guia, vários estudos têm utilizado telas de alta produtividade, juntamente com a próxima geração sequenciamento para compilar recursos de sequência de guias bem sucedida47,79,,93,94, 95,96. Esses recursos têm sido utilizados para desenvolver algoritmos preditivos e ferramentas online para ajudar na guia seleção44,,45,46,47,,48. Tais algoritmos baseiam-se em telas usando sistemas baseados em DNA para guia de expressão de RNA. Guias são expressos usando um promotor Pol III, e sua expressão é, portanto, propenso às limitações associadas com transcrição Pol III, tais como o encerramento prematuro quando encontrar faixas de uracil97,98, 99. No entanto, o uso de RNPs feito com in vitro-guia sintetizado RNAs ignora essas preocupações e simplifica as restrições no design de guia. Uma característica comum que surgiu a partir destes algoritmos e foi confirmada em inúmeros estudos com edição de genoma altamente eficaz, é a presença de uma purina, particularmente uma guanina, na extremidade 3 ' da sequência de destino-específico do guia. Esse recurso de guia tem tido muito sucesso entre os organismos que variam de mamíferos a c. elegans, moscas da fruta e do zebrafish65,100,101. Além disso, para o c. elegans, criar guias com um dinucleótido GG na extremidade 3 ' da região alvo do guia é uma estratégia eficaz para a estimativa de RNAs altamente eficaz guia65. Idealmente, teste vários guias em paralelo para determinar qual é o mais bem sucedido para um determinado aplicativo.
Ao tentar introduzir uma sequência de ADN no genoma, o design do doador ou modelo DNA também é crucial. Doadores do oligonucleotide single-stranded (ssODNs) são inseridos mais confiável do que outros modelos de reparo típico, double-stranded linear e10255,54,do DNA do plasmídeo. Em alguns loci, HDR eficiência pode ser melhorada com ssODNs que são complementares para o não-alvo ou deslocados de uma cadeia de DNA e possuir armas de homologia que são assimétricas no comprimento27,55. Desde que o modelo de reparação está sendo inserido no local do corte e inclui a sequência alvo, deve tomar medidas para impedir que Cas9 fendendo o doador de DNA antes ou após a inserção de genômica. Isso é conseguido fazendo mutações silenciosas para a sequência de PAM ou a região de semente, evitando o reconhecimento por Cas9 mantendo a função do gene inserido21,103. Embora ainda único nucleotídeo altera para o PAM são susceptíveis de abolir a vinculação104, tentar mudar pelo menos quatro nucleotídeos para ser seguro.
Significado e aplicações futuras:
Genoma de edição com CRISPR-Cas9 tem emergido como um método poderoso permitindo fácil manipulação genética de qualquer organismo. Edição com a RNP Cas9 leva um pouco mais de esforço no início, mas é simples de usar, uma vez que os reagentes e os protocolos são estabelecidos em um laboratório. Edição de células com RNP pré-montados em vez de Plasmídeo conduz a maior eficiência de edição global, incluindo a inserção de gene difícil de atingir através de HDR, com menos efeitos fora do alvo24,25,26 , 27 , 29. Além disso, experimentadores evitar problemas com a expressão de gene, RNA degradação, enrolamento de proteínas e a associação entre gRNA e Cas9 moléculas sintetizadas separadamente dentro da célula de22,23. Edição de RNP também contorna a preocupações de segurança sobre mutagênese insercional e expressão sustentado que pode surgir quando os métodos de entrega virais são utilizados clinicamente14. Devido a estas vantagens, muitos cientistas realizando pré-clínicos, experimentos de prova de conceito favorecem a RNP edição para aplicações terapêuticas em seres humanos. Abordagens de edição baseada em RNP genoma tanto in vivo e ex vivo estão em desenvolvimento para tratar ou até mesmo curar uma variedade de condições, de doenças genéticas como Duchenne muscular dystrophy105 e doença falciforme-27 HIV29 e câncer11. Curiosamente, a RNP Cas9 cada vez mais é empregado como um método de entrega na engenharia agrícola, pois permite que o ''DNA-livre edição de plantas33,34,36.
Disclosures
Os autores Alexander Marson e Jacob E. Corn são co-fundadores da terapêutica de holofotes. Jacob E. Corn é um conselheiro para missão terapêutica e seu laboratório recebeu apoio de pesquisa patrocinados da Pfizer e da AstraZeneca. Alexander Marson é um conselheiro para terapêutica de Juno e terapêutica do Pacto, e seu laboratório recebeu apoio de pesquisa patrocinados da terapêutica de Juno, Epinomics e Sanofi. Seu laboratório também solicitou patentes relacionadas com tecnologia Cas9 RNP.
Acknowledgments
Agradecemos muitos membros anteriores dos nossos laboratórios e da comunidade de edição de genoma de Bay Area por suas contribuições para o desenvolvimento desses métodos. Agradecemos Ross Wilson criticamente lendo este manuscrito.
Pesquisa de Alexander Marson é suportada por um presente do Jake Aronov e conceder de uma sociedade nacional de esclerose múltipla (CA. 1074-A-21). Alexander Marson detém um prêmio de carreira para médicos cientistas da Burroughs Wellcome fundo e um investigador de Biohub Chan Zuckerberg. Pesquisa de Jacob E. Corn é suportada pelo Instituto da Califórnia, a herança médica Instituto de investigação médica e o Li Ka Shing Foundation para medicina regenerativa. Pesquisa Behnom Farboud e de Barbara J. Meyer é financiada em parte pela subvenção NIGMS R01 GM030702 para Barbara J. Meyer, que é um investigador do Howard Hughes Medical Institute. Pesquisa Erin Jarvis e de Nipam H. Patel é financiada em parte pela subvenção NSF 1257379-IOS e Erin Jarvis reconhece o suporte de uma GRFP NSF e uma bolsa de pós-graduação Philomathia.
Materials
| Name | Company | Catalog Number | Comments |
| Reagents/Materials | |||
| DNA oligonucleotides | Integrated DNA Technologies | - | IDT will provide custom DNA sequences, including those in Table 1 |
| Guide RNAs | Synthego | - | Synthego will provide high-quality sgRNAs for S. pyogenes Cas9, including custom sgRNAs containing the targeting sequences included in Table 1 |
| Purified Cas9 protein (EnGen Cas9 NLS, S. pyogenes) | New England Biosciences | M0646T | If possible, purifying Cas9 in-house or purchasing from local core facilities is a less expensive option |
| Normal peripheral blood CD34+ stem/progenitor cells | AllCells | PB032-2 | |
| StemSpan SFEM | StemCell Technologies | 09650 | |
| StemSpan CC110 | StemCell Technologies | 02697 | |
| P3 Primary Cell 4D-Nucleofector X Kit | Lonza | V4XP-3032 | |
| RPMI-1640 Medium, With sodium bicarbonate, without L-glutamine, liquid | Sigma | R0883-6X500ML | |
| EasySep™ Human T Cell Isolation Kit | Stemcell | 17951 | |
| cell culture plate, 96 wells, round | Fisher Scientific | 3799 | |
| CTS (Cell Therapy Systems) Dynabeads CD3/CD28 | Life Tech | 40203D | |
| Reombinant Human IL-2 | UCSF Pharmacy | NA | |
| SepMate-50 500-pack IVD | Stemcell Technologies | 85460 | |
| OP50 Escherichia coli | Caenorhabditis Genetics Center | OP-50 | https://cgc.umn.edu/ |
| Nematode Growth Media agar in petri dishes | - | - | See Stiernagle, T (ref. 59) |
| Standard borosilicate glass capillaries with filament: 4 in (100 mm), 1/0.58 OD/ID | World Precision Instruments | 1B100F-4 | |
| Single-barrel standard borosilicate glass capillaries: 6 in (152 mm), 2/1.12 OD/ID | World Precision Instruments | 1B200-6 | |
| Cover glass; 24 × 50 mm | Thermo Fisher Scientific | 12-544E | |
| Cover glass; 22 × 22 mm | Thermo Fisher Scientific | 12-518-105K | |
| Apex LE agarose | Genesee Scientific | 20-102 | |
| Halocarbon oil 700 | Sigma-Aldrich | H8898-100ML | |
| pCFJ90 plasmid | Addgene | 19327 | |
| Compressed nitrogen | - | ||
| 60 mM culture dishes | BD | ||
| Capillary tubes with filament: 4 in (1.0 mm) | World Precision Instruments | T2100F-4 | |
| Sylgard 184 | Dow Corning | ||
| Petri dishes (100 × 15 mm) | - | ||
| Tungsten wire (0.005 in. diameter) | Ted Pella | ||
| Perfluoroalkoxy alkane (PFA) | - | ||
| Marine salt | - | ||
| 9" pasteur pipettes | - | ||
| Phenol red | - | ||
| Nuclease-free water | - | ||
| Equipment | |||
| 4D Nucleofector | Lonza | AAF-1002X | |
| MZ75 Stereomicroscope | Leica | Out-of-production. Current model is the M80 Stereomicroscope | |
| Axio Vert35 inverted phase contrast fluorescent microscope | Zeiss | Out-of-production. Current model is the Axio VertA.1 | |
| Laser-based micropipette puller (for C. elegans protocol) | Sutter Instrument | FG-P2000 | |
| Picoliter Microinjector (for C. elegans protocol) | Warner Instruments | PLI-100A | |
| Three-axis Joystick oil hydraulic micromanipulator | Narishige International | MO-202U | |
| Coarse manipulator | Narishige International | MMN-1 | |
| Micropipette puller (for P. hawaiensis protocol) | Sutter Instrument | P-80/PC | |
| Microinjector (for P. hawaiensis protocol) | Narishige | IM300 | |
| Microloader pipette tips | Eppendorf | 5242956003 | |
| NG-agar |
References
- Komor, A. C., Badran, A. H., Liu, D. R. CRISPR-based technologies for the manipulation of eukaryotic genomes. Cell. , 1-17 (2016).
- Barrangou, R., Horvath, P. A decade of discovery: CRISPR functions and applications. Nature Microbiology. 2, 1-9 (2017).
- Martin, A., Serano, J. M., et al. CRISPR/Cas9 mutagenesis reveals versatile roles of Hox genes in crustacean limb specification and evolution. Current Biology. 26 (1), 14-26 (2016).
- Goldstein, B., King, N. The future of cell biology: emerging model organisms. Trends in Cell Biology. 26 (11), 818-824 (2016).
- Mali, P., Yang, L., et al. RNA-guided human genome engineering via Cas9. Science. 339 (6121), 823-826 (2013).
- Cong, L., Ran, F. A., et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 339 (6121), 819-823 (2013).
- Deltcheva, E., Chylinski, K., et al. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature. 471 (7340), 602-607 (2011).
- Jinek, M., Chylinski, K., Fonfara, I., Hauer, M., Doudna, J. A., Charpentier, E. A Programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 337 (6096), 816-821 (2012).
- Nowak, C. M., Lawson, S., Zerez, M., Bleris, L. Guide RNA engineering for versatile Cas9 functionality. Nucleic Acids Research. 44 (20), 9555-9564 (2016).
- Jiang, W., Cox, D., Zhang, F., Bikard, D., Marraffini, L. A. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nature Biotechnology. , 1-9 (2013).
- Rupp, L. J., Schumann, K., et al. CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Scientific Reports. 7 (1), 737 (2017).
- Graham, D. B., Root, D. E. Resources for the design of CRISPR gene editing experiments. Genome Biology. 16, 260 (2015).
- Wang, H., La Russa, M., Qi, L. S. CRISPR/Cas9 in genome editing and beyond. Annual Review of Biochemistry. 85, 2270 (2016).
- Nelson, C. E., Gersbach, C. A. Engineering delivery vehicles for genome editing. Annual Review of Chemical and Biomolecular Engineering. 7, 637-662 (2016).
- Yin, H., Kauffman, K. J., Anderson, D. G. Delivery technologies for genome editing. Nature Reviews Drug Discovery. 16 (6), 387-399 (2017).
- Zuris, J. A., Thompson, D. B., et al. Cationic lipid-mediated delivery of proteins enables efficient protein-based genome editing in vitro and in vivo. Nature Biotechnology. 33 (1), 73-80 (2015).
- Cho, S. W., Lee, J., Carroll, D., Kim, J. -S., Lee, J. Heritable gene knockout in Caenorhabditis elegans by direct injection of Cas9-sgRNA ribonucleoproteins. Genetics. 195 (3), 1177-1180 (2013).
- Wang, W., Kutny, P. M., et al. Delivery of Cas9 protein into mouse zygotes through a series of electroporation dramatically increases the efficiency of model creation. Journal of Genetics and Genomics. 43 (5), 319-327 (2016).
- Chen, S., Lee, B., Lee, A. Y. -F., Modzelewski, A. J., He, L. Highly efficient mouse genome editing by CRISPR ribonucleoprotein electroporation of zygotes. Journal of Biological Chemistry. 291 (28), 14457-14467 (2016).
- Remy, S., Chenouard, V., et al. Generation of gene-edited rats by delivery of CRISPR/Cas9 protein and donor DNA into intact zygotes using electroporation. Scientific Reports. 7 (1), 16554 (2017).
- DeWitt, M. A., Corn, J. E., Carroll, D. Genome editing via delivery of Cas9 ribonucleoprotein. Methods. , 1-7 (2017).
- Hendel, A., Bak, R. O., et al. Chemically modified guide RNAs enhance CRISPR-Cas genome editing in human primary cells. Nature Biotechnology. 33 (9), 985-989 (2015).
- Thyme, S. B., Akhmetova, L., Montague, T. G., Valen, E., Schier, A. F. Internal guide RNA interactions interfere with Cas9-mediated cleavage. Nature Communications. 7, 11750 (2016).
- Kim, S., Kim, D., Cho, S. W., Kim, J., Kim, J. -S. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Research. 24 (6), 1012-1019 (2014).
- Lin, S., Staahl, B. T., Alla, R. K., Doudna, J. A. Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. eLife. 3, 04766 (2014).
- Liang, X., Potter, J., et al. Rapid and highly efficient mammalian cell engineering via Cas9 protein transfection. Journal of Biotechnology. 208, 44-53 (2015).
- DeWitt, M. A., Magis, W., Bray, N. L., Wang, T. Selection-free genome editing of the sickle mutation in human adult hematopoietic stem/progenitor cells. Science Translational Medicine. 8 (360), (2016).
- Ramakrishna, S., Kwaku Dad, A. -B., Beloor, J., Gopalappa, R., Lee, S. -K., Kim, H. Gene disruption by cell-penetrating peptide-mediated delivery of Cas9 protein and guide RNA. Genome Research. 24 (6), 1020-1027 (2014).
- Schumann, K., Lin, S., et al. Generation of knock-in primary human T cells using Cas9 ribonucleoproteins. Proceedings of the National Academy of Sciences of the United States of America. 112 (33), 10437-10442 (2015).
- Lee, J. -S., Kwak, S. -J., et al. RNA-guided genome editing in Drosophila with the purified Cas9 protein. G3: Genes, Genomes, Genetics (Bethesda, MD). 4 (7), 1291-1295 (2014).
- Sung, Y. H., Kim, J. M., et al. Highly efficient gene knockout in mice and zebrafish with RNA-guided endonucleases. Genome Research. 24 (1), 125-131 (2014).
- Menoret, S., De Cian, A., et al. Homology-directed repair in rodent zygotes using Cas9 and TALEN engineered proteins. Scientific Reports. 5, 14410 (2015).
- Woo, J. W., Kim, J., et al. DNA-free genome editing in plants with preassembled CRISPR-Cas9 ribonucleoproteins. Nature Biotechnology. 33 (11), 1162-1164 (2015).
- Malnoy, M., Viola, R., et al. DNA-free genetically edited grapevine and apple protoplast using CRISPR/Cas9 ribonucleoproteins. Frontiers in Plant Science. 7, 1904 (2016).
- Svitashev, S., Schwartz, C., Lenderts, B., Young, J. K., Mark Cigan, A. Genome editing in maize directed by CRISPR-Cas9 ribonucleoprotein complexes. Nature Communications. 7, 13274 (2016).
- Liang, Z., Chen, K., et al. Efficient DNA-free genome editing of bread wheat using CRISPR/Cas9 ribonucleoprotein complexes. Nature Communications. 8, 14261 (2017).
- Shin, S. -E., Lim, J. -M., et al. CRISPR/Cas9-induced knockout and knock-in mutations in Chlamydomonas reinhardtii. Scientific Reports. 6, 27810 (2016).
- Pohl, C., Kiel, J. A. K. W., Driessen, A. J. M., Bovenberg, R. A. L., Nygård, Y. CRISPR/Cas9 based genome editing of Penicillium chrysogenum. ACS Synthetic Biology. 5 (7), 754-764 (2016).
- Grahl, N., Demers, E. G., Crocker, A. W., Hogan, D. A. Use of RNA-protein complexes for genome editing in non-albicans Candida species. mSphere. 2 (3), (2017).
- Rivera-Torres, N., Kmiec, E. B. A standard methodology to examine on-site mutagenicity as a function of point mutation repair catalyzed by CRISPR/Cas9 and ssODN in human cells. Journal of Visualized Experiments. (126), (2017).
- Nandal, A., Mallon, B., Telugu, B. P. Efficient generation and editing of feeder-free IPSCs from human pancreatic cells using the CRISPR-Cas9 system. Journal of Visualized Experiments. (129), (2017).
- Mohr, S. E., Hu, Y., Ewen-Campen, B., Housden, B. E., Viswanatha, R., Perrimon, N. CRISPR guide RNA design for research applications. The FEBS Journal. 283 (17), 3232-3238 (2016).
- Bauer, D. E., Canver, M. C., Orkin, S. H. Generation of genomic deletions in mammalian cell lines via CRISPR/Cas9. Journal of Visualized Experiments. (95), e52118 (2015).
- Hsu, P. D., Scott, D. A., et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nature Biotechnology. 31 (9), 827-832 (2013).
- Heigwer, F., Kerr, G., Boutros, M. E-CRISP: fast CRISPR target site identification. Nature Methods. 11 (2), 122-123 (2014).
- Moreno-Mateos, M. A., Vejnar, C. E., et al. CRISPRscan: designing highly efficient sgRNAs for CRISPR-Cas9 targeting in vivo. Nature Methods. 12 (10), 982-988 (2015).
- Labun, K., Montague, T. G., Gagnon, J. A., Thyme, S. B., Valen, E. CHOPCHOP v2: a web tool for the next generation of CRISPR genome engineering. Nucleic Acids Research. 44, 272-276 (2016).
- Haeussler, M., Schönig, K., et al. Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biology. 17 (1), 148 (2016).
- Lo, T. -W., Pickle, C. S., et al. Precise and heritable genome editing in evolutionarily diverse nematodes using TALENs and CRISPR/Cas9 to engineer insertions and deletions. Genetics. 195 (2), 331-348 (2013).
- Bassett, A., Liu, J. -L. CRISPR/Cas9 mediated genome engineering in Drosophila. Methods. 69 (2), 128-136 (2014).
- Prior, H., Jawad, A. K., MacConnachie, L., Beg, A. A. Highly efficient, rapid and co-CRISPR independent genome editing in Caenorhabditis elegans. G3: Genes, Genomes, Genetics. , Bethesda, MD. (2017).
- Hirsh, A. Cas9 expression and purification protocol. protocols.io. , (2017).
- DeWitt, M. A., Wong, J. In vitro transcription of guide RNAs. protocols.io. , (2017).
- Yang, L., Guell, M., et al. Optimization of scarless human stem cell genome editing. Nucleic Acids Research. 41 (19), 9049-9061 (2013).
- Richardson, C. D., Ray, G. J., DeWitt, M. A., Curie, G. L., Corn, J. E. Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA. Nature Biotechnology. 34 (3), 339-344 (2016).
- Paix, A., Folkmann, A., Seydoux, G. Precision genome editing using CRISPR-Cas9 and linear repair templates in C. elegans. Methods. 121-122, 86-93 (2017).
- Mello, C., Fire, A. DNA transformation. Methods in Cell Biology. 48, 451-482 (1995).
- Sutter Pipette Cookbook. , Available from: https://www.sutter.com/PDFs/pipette_cookbook.pdf (2017).
- Stiernagle, T. Maintenance of C. elegans. WormBook: the online review of C. elegans biology. , (2006).
- Evans, T. C. Transformation and microinjection. WormBook: the online review of C. elegans biology. , (2006).
- Berkowitz, L. A., Knight, A. L., Caldwell, G. A., Caldwell, K. A. Generation of stable transgenic C. elegans using microinjection. Journal of Visualized Experiments. (18), (2008).
- Kim, H., Ishidate, T., et al. A co-CRISPR strategy for efficient genome editing in Caenorhabditis elegans. Genetics. 197 (4), 1069-1080 (2014).
- Arribere, J. A., Bell, R. T., Fu, B. X. H., Artiles, K. L., Hartman, P. S., Fire, A. Z. Efficient marker-free recovery of custom genetic modifications with CRISPR/Cas9 in Caenorhabditis elegans. Genetics. 198 (3), 837-846 (2014).
- Ward, J. D. Rapid and precise engineering of the Caenorhabditis elegans genome with lethal mutation co-conversion and inactivation of NHEJ repair. Genetics. 199 (2), 363-377 (2015).
- Farboud, B., Meyer, B. J. Dramatic enhancement of genome editing by CRISPR/Cas9 through improved guide RNA design. Genetics. 199 (4), 959-971 (2015).
- Wood, A. J., Lo, T. -W., et al. Targeted genome editing across species using ZFNs and TALENs. Science. 333 (6040), 307 (2011).
- Friedland, A. E., Tzur, Y. B., Esvelt, K. M., Colaiácovo, M. P., Church, G. M., Calarco, J. A. Heritable genome editing in C. elegans via a CRISPR-Cas9 system. Nature Methods. 10 (8), 741-743 (2013).
- Dickinson, D. J., Ward, J. D., Reiner, D. J., Goldstein, B. Engineering the Caenorhabditis elegans genome using Cas9-triggered homologous recombination. Nature Methods. 10 (10), 1028-1034 (2013).
- Rehm, E. J., Hannibal, R. L., Chaw, R. C., Vargas-Vila, M. A., Patel, N. H. Injection of Parhyale hawaiensis blastomeres with fluorescently labeled tracers. Cold Spring Harbor Protocols. 2009 (1), (2009).
- Gerberding, M., Browne, W. E., Patel, N. H. Cell lineage analysis of the amphipod crustacean Parhyale hawaiensis reveals an early restriction of cell fates. Development (Cambridge, England). 129 (24), 5789-5801 (2002).
- Browne, W. E., Price, A. L., Gerberding, M., Patel, N. H. Stages of embryonic development in the amphipod crustacean, Parhyale hawaiensis. Genesis. 42 (3), New York, NY. 124-149 (2005).
- Rehm, E. J., Hannibal, R. L., Chaw, R. C., Vargas-Vila, M. A., Patel, N. H. Fixation and dissection of Parhyale hawaiensis embryos. Cold Spring Harbor Protocols. 2009 (1), (2009).
- Rehm, E. J., Hannibal, R. L., Chaw, R. C., Vargas-Vila, M. A., Patel, N. H. In situ hybridization of labeled RNA probes to fixed Parhyale hawaiensis embryos. Cold Spring Harbor Protocols. 2009 (1), (2009).
- Rehm, E. J., Hannibal, R. L., Chaw, R. C., Vargas-Vila, M. A., Patel, N. H. Antibody staining of Parhyale hawaiensis embryos. Cold Spring Harbor Protocols. 2009 (1), (2009).
- Kontarakis, Z., Pavlopoulos, A. Transgenesis in non-model organisms: the case of Parhyale. Methods in Molecular Biology. 1196, Clifton, NJ. 145-181 (2014).
- Kim, H. J., Lee, H. J., Kim, H., Cho, S. W., Kim, J. -S. Targeted genome editing in human cells with zinc finger nucleases constructed via modular assembly. Genome Research. 19 (7), 1279-1288 (2009).
- Qiu, P., Shandilya, H., D'Alessio, J. M., O'Connor, K., Durocher, J., Gerard, G. F. Mutation detection using Surveyor nuclease. BioTechniques. 36 (4), 702-707 (2004).
- Brinkman, E. K., Chen, T., Amendola, M., van Steensel, B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Research. 42 (22), 168 (2014).
- Tsai, S. Q., Zheng, Z., et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nature Biotechnology. 33 (2), 187-197 (2015).
- Frock, R. L., Hu, J., Meyers, R. M., Ho, Y. -J., Kii, E., Alt, F. W. Genome-wide detection of DNA double-stranded breaks induced by engineered nucleases. Nature Biotechnology. 33 (2), 179-186 (2015).
- Smith, C., Gore, A., et al. Whole-genome sequencing analysis reveals high specificity of CRISPR/Cas9 and TALEN-based genome editing in human iPSCs. Cell Stem Cell. 15 (1), 12-13 (2014).
- Veres, A., Gosis, B. S., et al. Low incidence of off-target mutations in individual CRISPR-Cas9 and TALEN targeted human stem cell clones detected by whole-genome sequencing. Cell Stem Cell. 15 (1), 27-30 (2014).
- Kim, D., Bae, S., et al. Digenome-seq: genome-wide profiling of CRISPR-Cas9 off-target effects in human cells. Nature Methods. 12 (3), 237-243 (2015).
- Hendel, A., Fine, E. J., Bao, G., Porteus, M. H. Quantifying on- and off-target genome editing. Trends in Biotechnology. 33 (2), 132-140 (2015).
- O'Geen, H., Yu, A. S., Segal, D. J. How specific is CRISPR/Cas9 really. Current Opinion in Chemical Biology. 29, 72-78 (2015).
- Tsai, S. Q., Joung, J. K. Defining and improving the genome-wide specificities of CRISPR-Cas9 nucleases. Nature Reviews Genetics. 17 (5), 300-312 (2016).
- Hoban, M. D., Cost, G. J., et al. Correction of the sickle cell disease mutation in human hematopoietic stem/progenitor cells. Blood. 125 (17), 2597-2604 (2015).
- Simeonov, D. R., Gowen, B. G., et al. Discovery of stimulation-responsive immune enhancers with CRISPR activation. Nature. , (2017).
- Hultquist, J. F., Schumann, K., et al. A Cas9 ribonucleoprotein platform for functional genetic studies of HIV-host interactions in primary human T cells. Cell Reports. 17 (5), 1438-1452 (2016).
- Paix, A., Wang, Y., et al. Scalable and versatile genome editing using linear DNAs with microhomology to Cas9 sites in Caenorhabditis elegans. Genetics. 198 (4), 1347-1356 (2014).
- Lee, K., Mackley, V. A., et al. Synthetically modified guide RNA and donor DNA are a versatile platform for CRISPR-Cas9 engineering. eLife. 6, (2017).
- Minkenberg, B., Wheatley, M., Yang, Y. CRISPR/Cas9-enabled multiplex genome editing and its application. Progress in Molecular Biology and Translational Science. 149, 111-132 (2017).
- Doench, J. G., Fusi, N., et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nature Biotechnology. 34 (2), 184-191 (2016).
- Doench, J. G., Hartenian, E., et al. Rational design of highly active sgRNAs for CRISPR-Cas9-mediated gene inactivation. Nature Biotechnology. , 1-8 (2014).
- Liu, H., Wei, Z., Dominguez, A., Li, Y., Wang, X., Qi, L. S. CRISPR-ERA: a comprehensive design tool for CRISPR-mediated gene editing, repression and activation. Bioinformatics (Oxford, England). 31 (22), 3676-3678 (2015).
- Wu, X., Scott, D. A., et al. Genome-wide binding of the CRISPR endonuclease Cas9 in mammalian cells. Nature Biotechnology. 32 (7), 670-676 (2014).
- Bogenhagen, D. F., Brown, D. D. Nucleotide sequences in Xenopus 5S DNA required for transcription termination. Cell. 24 (1), 261-270 (1981).
- Cozzarelli, N. R., Gerrard, S. P., Schlissel, M., Brown, D. D., Bogenhagen, D. F. Purified RNA polymerase III accurately and efficiently terminates transcription of 5S RNA genes. Cell. 34 (3), 829-835 (1983).
- Chen, B., Gilbert, L. A., et al. Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell. 155 (7), 1479-1491 (2013).
- Gagnon, J. A., Valen, E., et al. Efficient mutagenesis by Cas9 protein-mediated oligonucleotide insertion and large-scale assessment of single-guide RNAs. PLoS ONE. 9 (5), 98186 (2014).
- Ren, X., Yang, Z., et al. Enhanced specificity and efficiency of the CRISPR/Cas9 system with optimized sgRNA parameters in Drosophila. Cell Reports. 9 (3), 1151-1162 (2014).
- Ran, F. A., Hsu, P. D., Wright, J., Agarwala, V., Scott, D. A., Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nature Protocols. 8 (11), 2281-2308 (2013).
- Serano, J. M., Martin, A., et al. Comprehensive analysis of Hox gene expression in the amphipod crustacean Parhyale hawaiensis. Developmental Biology. 409 (1), 297-309 (2016).
- Sternberg, S. H., Redding, S., Jinek, M., Greene, E. C., Doudna, J. A. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature. , 1-17 (2014).
- Lee, K., Conboy, M., et al. Nanoparticle delivery of Cas9 ribonucleoprotein and donor DNA in vivo induces homology-directed DNA repair. Nature Biomedical Engineering. 1 (11), 889-901 (2017).
