

Summary
소 Cas9를 활용 하 여 복잡 한 ribonucleoprotein (RNP)은 정확 하 고 효율적인 게놈 편집에 대 한 강력한 방법입니다. 여기, 우리가 강조의 유틸리티 세포와 생물의 광범위 한 범위에 걸쳐 기본 인간 세포 등 모두 클래식 하 고 모형 유기 체를 신흥.
Abstract
사이트별 진 핵 게놈 편집 CRISPR (클러스터 정기적으로 interspaced 짧은 구조 반복)-Ca (CRISPR 관련 된) 시스템 신속 하 게 다양 한 생물 학적 질문을 추구 하는 연구원 사이 평범한 되고있다. 사용자는 가장 자주 쉽게 재설정된 가이드 RNA (gRNA)와 함께 복잡 한 연쇄 상 구 균 pyogenes 에서 파생 하는 Cas9 단백질을 사용 합니다. 이러한 구성 요소는 셀에 도입 하 고 이중 가닥 DNA (dsDNA) 게놈의 상호 보완적인 영역을 페어링 자료를 통해 효소 앞 두 배 물가 틈 (DSB)를 생성 하기 위해 두 가닥. 후속 복구 무작위 삽입 또는 삭제 이벤트 (indels) 또는 휴식의 사이트에서 실험 제공 DNA의 이끌어 낸다.
순화 된 단일 가이드 RNA와 Cas9 단백질, 소는 RNP 형성 하 고 세포에 직접 전달의 사용은 매우 효율적인 유전자 편집 달성 하기 위한 강력한 접근 이다. RNP 편집 특히 유전자 삽입, 결과 달성 도전 자주 그의 속도를 향상 시킵니다. 셀 내에 Cas9 RNP의 짧은 지 속성은 플라스 미드를 통해 배달에 비해 적은 오프 대상 이벤트 리드.
자사의 장점에도 불구 하 고 CRISPR 유전자 편집의 많은 캐주얼 사용자 덜이 기술을 잘 알고 있다. 낮은 진입 장벽, 우리는 그것의 독특한 혜택 및 다양 한 응용 프로그램 컨텍스트 범위에서 RNP 전략을 구현 하기 위한 상세한 프로토콜 개요. 우리는 두 가지 유형의 기본 인간 세포, T 세포 및 조 혈 줄기/시 조 세포 (HSPCs)에서 편집 커버. 우리는 또한 어떻게 편집 하면 Cas9 RNP 최근 클래식 모델 거 위 꼬마 선 충 그리고 더 많은 포함 한 전체 유기 체의 손쉬운 유전자 조작 도입 모델 갑각류, Parhyale hawaiensis을 보여줍니다.
Introduction
fThe CRISPR Cas9 시스템 과학자 들은 게놈1의 대상된 영역을 변경할 수 있습니다. 이 신속 하 고 저렴 한 기술 기초 연구에 혁명을 고2넘어 개인된 질병 치료, 정밀 농업의 발전에 지대한 영향을 만들 것을 약속 드립니다. CRISPR 편집 democratizing 도구 이며 게놈 엔지니어링, 그냥 기본적인 분자 생물학 기술 없이 특정 전문 지식이 필요로 새로운 실험실에는 시스템을 구현 합니다. 연구원은 지금 이전 다루기 힘든 생물 유전자 조작3,4에 대 한 몇 가지 대체 수단을 공부할 수 있습니다. 지난 5 년간 혼자 CRISPR 게놈 편집 사용 되었습니다 200 개 이상의 다른 척추 동물, 무척추동물, 식물, 그리고 미생물 종 하.
CRISPR 간결한 방어 통로에서 적응, 사이트별 게놈 편집에 필요한 핵심 요소 Cas9 단백질의 S. pyogenes 에서 일반적으로 및 추가 핵 지 방화 신호 (NLS), 그리고 그것의 전문화 된 코 돈 최적화 RNA 가이드5,6. 여기 설명 하지, 비록 다른 Cas9 orthologues 또는 CRISPR endonucleases 또한 사용할 수 있습니다. 자연스럽 게 발생 gRNA 두 별도로 베낀된 조각, CRISPR RNA (crRNA) 및 트랜스 활성화로 구성 된다 crRNA (tracrRNA)7. 이러한 RNAs 단일 가이드 RNA (sgRNA)8로 알려진 단일 사본으로 융합 될 수 있다. 대부분 게놈 편집자 선택 유선형된 sgRNA9, 듀얼 가이드 이기도 하지만10,11을 정기적으로 사용. 경험 20-뉴클레오티드 (nt) 게놈 DNA 대상, 그것은 protospacer 인접 한 모티브 (PAM), 라는 Cas9 인식에 필요한 짧은 라이센스 서명 옆 거짓말 보장 선택한 보완 시퀀스12 포함 하는 gRNA 디자인 .
일단 셀, 안으로 RNP 복잡 한 게놈 목표, gRNA 기초 쌍 무료 dna 물가, 및 다음 효소 앞 이중 가닥을 생성 하기 위해 두 DNA 가닥 휴식2. 두 개 이상의 경로 중 하나에 의해 DSB를 해결 하는 세포 수리 기계: 오류가 아닌-동종 끝 결합 (NHEJ) 통로 또는 상 동 감독 수리 (HDR)는 완벽 하 게 틈의 양쪽에 상 동의 ' 무기'를 포함 하는 DNA를 통합. 이전 복구 경로 일반적으로 이끌어 낸다 indel 형성과 필연적인 유전자 장애, 후자 허용 경험을 삽입 하거나 DNA 시퀀스1을 변경 하는 동안.
편집 효율성과 정확도는 Cas9와 gRNA는 셀에 입력 하는 방법에 따라 달라 집니다. 이러한 구성 요소는 배양된 세포, 배아, 또는 핵 산의 모양으로 또는 유기 체 소 RNP 복잡 한13,,1415로 전달 수 있습니다. 일반적인 핵 산 기반 배달 방법에는 바이러스 성 변환, transfection, 또는 electroporation mRNA 또는 플라스 미드 DNA의 포함 됩니다. Cas9 단백질 및 가이드 RNA는 세포 내에서 생산 다음 및 그들은 복합물을 형성 하.
RNP의 직접 배달 Cas9 단백질 및 가이드 RNA의 별도 정화를 요구 한다. 이 사내, 행 해질 수 있다 또는 단백질과 sgRNA 여러 상용 공급 업체 중 하나에서 구입하실 수 있습니다. 획득 되 면 Cas9와 gRNA 효소 유능한 RNP 복합물을 형성 하기 위하여 섞는 그리고 수정 된 달걀/배아, 지질에 기초를 둔 transfection16또는 electroporation에 직접 주입 하 여 셀 소개. RNP 편집의 첫 번째 보고서 관련 C. 선 충 의 생식 선17에 주입. Microinjection은 여전히 효과적인 electroporation 마우스18,19 와 쥐20 배아에서 입증 되었습니다 비록 배아와 전체 유기 체, RNP 도입의 선호 하는 수단. 우리는 직접 주입 RNP C. 선 충 의 생식 선 및 P. hawaiensis 배아에 대 한 프로토콜을 설명 하 고 특수 유형의 electroporation RNP 인간의 기본 셀을 편집할 때 제공 하는 것이 좋습니다. 이 방법은, nucleofection, 최적화 된 electroporation 프로그램 및 셀 형식 관련 솔루션을 포함 하 세포질 및 핵21입력 RNP 하며
게놈 RNP 편집 여러 가지 이점을 제공 합니다. 때문에 단백질 및 RNA 구성 요소 조립, 배달 하기 전에 품질을 보장 될 수 있는, RNP 편집 관련 핵 산 기반 배달 된 많은 함정을 피할 수 있습니다. 즉, Cas9 인코딩 DNA 주인 게놈으로 통합의 위험, mRNA는 결코 저하, 노출 그리고 vivo에서 gRNA 또는 단백질 식, 접는, 그리고 협회22,23문제 circumvents. 또한, 낮은 독성 및 플라스 미드 기반 식, 셀24,25,,2627안으로 RNP의 짧은 반감기의 결과 보다 훨씬 적은 오프 대상 이벤트 리드 RNP 사용 하 여.
마지막으로, RNP 편집 명백 하 게 이끌어 인간 세포 라인, 1 차 셀의 다양 한에서 높은 편집 속도 같은 섬유 아 세포, 배아 줄기 세포 (ESCs), 유도 만능 줄기 세포 (iSPCs), HSPCs, T 세포16,24, 25,26,27,,2829; 무척 추 동물을 포함 하 여 C. 선 충, P. hawaiensis및 과일 파리3,,1730; zebrafish, 쥐, 및 쥐31,32; 처럼 척 추가 있는 종 식물 종 등 애기, 담배, 상 추, 쌀, 포도, 애플, 옥수수, 밀33,,3435,36; 그리고 Chlamydomonas, 푸른 곰 팡이, 및에서 Candida 종37,,3839. Indel 형성의 주파수 RNP 플라스 미드 배달에 비해 사용 하 높을 수 있습니다 그리고 HDR 중재 DNA 삽입25,,2729달성 하기 쉬울 수 있다.
여기에 설명 된 프로토콜 Cas9 RNP 사용 하 고 다양 한 생물 학적 시스템40,41, 특히 일 수 없는 셀에에서 적용 하는 것은 간단를 효과적이 고, 쉽게 적응할 수 있는 기술 이다 그리고 정확한 유전자 조작에 대 한 확고 한 시스템 없이 유기 체에서. 우리는 디자인 가져오고 다른 모델 세포 유형 및 유기 체 그것의 사용을 취재 하기 전에 Cas9 RNP를 조립 하는 방법을 설명 하 여 시작 합니다. 조 혈 줄기/뿌리 (HSPCs) 세포 그리고 T 세포 그들은 함께 단계 2와 3이이 프로토콜의 덮여 있다 그래서 동일한 방법을, nucleofection를 사용 하 여 편집 됩니다. C에 대 한 절차를 편집. 선 충 4-5, 단계에 설명 되어 있습니다 및 P. hawaiensis 편집 단계 6과 7에에서 덮여 있다. 마지막으로, 모든 유기 체에서 유전자 편집 실험의 성공 유전자 시퀀싱에 의해 사정 될 수 있습니다, 모든 세포와 유기 체는 프로토콜에서 설명에 대 한 가능한 분석 방법을 설명 하는 하위 단계 8 단계에서 설명 되어 있습니다.
Protocol
1. RNP 어셈블리
-
사전에, 미리 모든 RNA, DNA, 단백질 구성 획득 실험을 디자인 합니다. 첫 번째 패스 표 1에 나열 된 긍정적인 컨트롤 중 하나를 시도 하 고 재료의 테이블에 설명 된 상업용 시 약을 사용 하 여 신뢰할 수 있는 실험 설계 및 자료의 무결성을 보장 합니다. 추가 도움말 새로운 게놈 편집 실험 계획에 대 한이 항목12,,4243에 논문을 참조.
참고: 일단 후속 단계에 설명 된 대로 조립, 사전에 준비 하는 RNPs 저장 될 수 있습니다-80 ° c.에- 대상에는 유전자를 선택한 후 사용 하 여 무료 온라인 도구 중 하나는 최적의 gRNA44,45,,4647,48디자인. 녹아웃 생성을 기대 하는 경우는 exon를 대상으로 해야 합니다.
참고: 이러한 도구는 인접 한 S. pyogenes PAM와 대상 사이트를 식별 하는 데 도움이 됩니다 시퀀스, 높은 품질 평가 점수, 그리고 낮은 떨어져 목표 점수. - 게시 방법8, S. pyogenes Cas9 단백질 정화 또는 상업용 공급 업체에서 구입.
- RNA 희석, RNP 준비, 및 단백질 저장, KCl, 10% 글리세롤의 1 mM의 TCEP HEPES pH 7.5, 150 m m의 20 밀리미터를 포함 하는 전형적인 Cas9 버퍼를 준비 합니다. 항상 nuclease 무료 물을 사용 하 여 버퍼 resuspend 또는 RNA 강 직을 방지 하기 위해 희석 하는 데 사용 됩니다.
- 생체 외에서 전사 게시 방법을 사용 하 여 통해 가이드 RNA (tracrRNA 및 crRNA 또는 sgRNA)을 생성 하거나는 핵 산 합성 회사17,,2149, 에서 구입 50 , 51.
- 유전자를 삽입 하는 경우 합성 하거나 구입할 기증자 DNA 템플렛.
- 단백질 및 RNA aliquots-80 ° C 및 해 동 즉시 사용 하기 전에 얼음에 저장 합니다.
참고: 각 동결-해 동 약간 효율성을 낮춘 다. 상세한, 오픈-액세스 프로토콜 Cas9 정화52 및 녹음 방송 생체 외에서 sgRNAs53 의 다른 곳에서 사용할 수 있습니다.
- 대상에는 유전자를 선택한 후 사용 하 여 무료 온라인 도구 중 하나는 최적의 gRNA44,45,,4647,48디자인. 녹아웃 생성을 기대 하는 경우는 exon를 대상으로 해야 합니다.
- C. 선 충과 일 경우 1.5 단계로 건너뜁니다. P. hawaiensis 프로토콜에 대 한 1.6 단계로 건너뜁니다. SgRNA를 사용 하는 경우 1.4 단계로 건너뜁니다. 1.3 1 차 셀 편집을 위한 gRNA 조립 단계를 진행 합니다.
-
TracrRNA와 crRNA의 아데닌 양을 혼합 하 여는 gRNA를 조립. 주식의 80 µ M gRNA, 약 50 게놈 편집 실험 100 µ L를 확인 합니다.
- 30 분 동안 37 ° C에서 gRNA를 품 어 고 천천히 실내 온도에 냉각.
-
RNP HSPC 및 T 셀 편집을 위한 준비: 10 µ L. 총 볼륨에 Cas9 단백질의 200 pmol에 gRNA의 1-2 x 어 금 니 금액을 매우 천천히 혼합 하 여 복잡 한 RNP 조립 추가 집중된 Cas9 gRNA (Cas9 버퍼에 미리 희석 해) 약 30 s 피 펫, 최종 Cas9 농도 20 µ M 데 려와 빠른 원형 만들기.
- Electroporation 큐 벳을 준비 합니다.
참고:이 프로토콜은 특정 테이블의 재료에 상업 시스템에 있지만 RNP 편집 또한 달성 될 수 있다 다른 electroporation 장치. - 각 베트 하 5 µ L (100 pmols, T 세포) 또는 RNP의 10 µ L (200 pmol, HSPCs)를 추가 합니다.
- 녹아웃, 보다 새로운 DNA를 삽입 큐 벳 또는 격판덮개의 우물 1 µ L 100 µ M (100 pmol) 단일 가닥 oligonucleotide 기증자 DNA (ssODN)25,,5455 의 추가.
- 1 차 셀 편집에 다음 지침에 대 한 2 단계를 건너뛰기 프로토콜.
- Electroporation 큐 벳을 준비 합니다.
-
C. 선 충 편집 RNP 준비: 20 µ L (최종 농도 괄호에서 주의 된다)의 최종 볼륨을 만들려면 다음과 같은 시 약을 추가 하 여 복잡 한 RNP 조립: Cas9 (2 µ M), HEPES pH 7.5 (10 µ M), KCl (115 µ M), crRNA (12 µ M) tracrRNA (40 µ M), 그리고는 수리 서식 파일이 필요한 경우 (0.5 µ M ssDNA 또는 350 ng / µ L dsDNA).
참고: Cas9 중재 DSB 템플릿 복구의 효율성은 dsDNA 수리 구조물;의 농도에 비례 따라서, 더 효율적인 복구 서식 파일의 높은 농도 템플릿 수리. 그러나, dsDNA의 µ 350 ng/L 보다 큰 포함 하는 혼합 주사 주입 된 벌레의 가능성을 줄이기 위해 표시 되었습니다. 따라서, 그것은,를 사용 하 여 dsDNA의 치 사 율을 최소화 하면서 복구 효율을 극대화 하기 위해 혼합에의 더 이상 350 ng / µ L 하지만 최고의.- 동시에 여러 개의 loci를 대상, 공동-CRISPR/co conversion 심사 방법 설명 필요에 따라 단계에서 5.4 여러 crRNAs를 추가 합니다. 하나 이상의 crRNA를 추가할 때는 각 마스터에 순차적으로 추가 합니다.
참고: 각 crRNA의 양이 필요가 없습니다 같은 것 하 고도 Cas9의 농도 변경 하지 않고 마스터 믹스에서 crRNAs의 총 농도 두배로 mutagenesis 특정 장소에서의 주파수 간섭에 표시 되지 않습니다. 예 페 외 에 자세하게 설명 되어 있습니다. 56. - Pipetting으로 혼합 하 고 회전 16000 x g 5에서 RNP 솔루션 s 솔루션은 튜브의 맨 아래에 수집 있는지 확인 하기.
- 15 m 37 ° C에서 솔루션을 품 어.
- 작은 얇은-지 루 microinjection 바늘을 방해할 수 있는 모든 입자를 1 분 동안 16000 x g에서 샘플 원심 상쾌한을 사용 하 여 후속 단계에.
- C. 선 충 프로토콜의 나머지 부분에 대 한 4 단계로 건너뜁니다.
- 동시에 여러 개의 loci를 대상, 공동-CRISPR/co conversion 심사 방법 설명 필요에 따라 단계에서 5.4 여러 crRNAs를 추가 합니다. 하나 이상의 crRNA를 추가할 때는 각 마스터에 순차적으로 추가 합니다.
-
P. hawaiensis 편집 RNP 준비: 단일 사용 Cas9 aliquots Cas9와 0.15% 페 놀 레드의 6.25 µ M의 최종 농도에 nuclease 무료 물과 페 놀 레드 (시각화 주사)에 대 한 그들을 diluting 하 여 준비.
- 2-5 gRNA Cas9 단백질 6 µ l. 추가 12 pmol Cas9의 gRNA, 2 µ M, 4 ~ 8 µ m, gRNA 농도 및 페 놀 레드 농도 0.05%를 최종 Cas9 농도 데리고 하의 전체 볼륨에의 어 금 니 과잉 배 혼합 하 여 복잡 한 RNP 조립.
- 복잡 일 분 동안 실내 온도에 혼합물은 RNP 품 어.
- P. hawaiensis 편집에 다음 지침에 대 한 6 단계 건너뛰기프로토콜.
2. 세포 문화 및 준비
참고: 생물학 안전 장은 2.1.1 3.3.3 단계를 수행 합니다.
-
구매 cryopreserved 인간 주변 혈액 CD34 동원+ HSPCs 공급 업체에서.
- 해 동 ~ 1 x106 HSPCs 37 ° C 물에 3 분 동안 목욕 하 고 15 mL 원뿔 튜브에 그들을 전송. 상업적인 소스에서 혈 청 무료 확장 매체의 10 mL를 추가 하 고 10 분은 상쾌한 제거 하 고 보충된 SFEM의 2 mL에 셀 resuspend 100 x g에서 혼합물을 회전. 6 잘 플레이트에 셀 접시 고 RNP electroporation 전에 24-48 h에 대 한 37 ° C 배양 기에서 문화.
- hemocytometer 셀을 계산 하 고 원심 분리기 튜브에 HSPCs 필요 (electroporated 하 베트 당 150000-200000 HSPCs)의 총 수를 전송.
- 작은 셀 10 분 x 100g에 튜브를 회전 합니다.
-
인간의 기본 CD4 구매 공급 업체에서+ T 세포 또는 조밀도 기온 변화도 원심 분리29에 의해 인간의 전체 혈액에서 그들을 분리.
- T 세포 활성화 전에 미리 αCD3 48-잘 문화 접시 코트 (UCHT1) 및 αCD28 (CD28.2). 10 µ g/mL αCD3 및 적어도 2 h 37 ° c.에 대 한 PBS에 10 µ g/mL αCD28 500 µ L로 플레이트 코트
참고: 일부 loci를 위한 NHEJ 달성 될 수 있다 사전 자극 없이 하지만이 단계를 포함 하 여 그것의 효율을 극대화. - ΑCD3/αCD28 항 체 바인딩 접시 [RPMI-1640 보충 L-글루타민, 상업 대안의 2 mM HEPES의 5 밀리미터를 가진 페니실린/스, 2-mercaptoethanol의 5 mM의 50 µ M의 50 µ g/mL RPMI 완전 한 매체에서에 37 ° C에서 48 h에 대 한 문화는 T 세포 비 본질적인 아미노산, 나트륨 pyruvate 그리고 10% (vol/vol) FBS의 5 mM]. 48-잘 접시의 당 미디어의 500 µ L에서 2000000 T 세포의 밀도에서 문화는 T 세포.
- Hemocytometer 및 전송 사용 하 여 개수는 T 세포는 electroporation에 필요한 T 세포의 총 수 원심 분리기 튜브 (electroporated 하 베트 당 100000-1000000 T 세포) 실험.
- 8 분 셀 작은 90 x g에 튜브를 회전 합니다. 만약 셀 밀도 그라데이션-2 일 이내 분리, 200 x g 8 분에 그들을 회전 합니다.
- T 세포 활성화 전에 미리 αCD3 48-잘 문화 접시 코트 (UCHT1) 및 αCD28 (CD28.2). 10 µ g/mL αCD3 및 적어도 2 h 37 ° c.에 대 한 PBS에 10 µ g/mL αCD28 500 µ L로 플레이트 코트
-
두 셀 형식에 대 한 피 펫/진공, 모든 거품을 제거와 함께 상쾌한 발음.
- 부드럽게 베트 당 20 µ L electroporation 버퍼의 셀 resuspend.
- 이미 잘 거품을 만들지 않고 아래로 pipetting으로 RNP, 그리고 믹스의 10 µ L를 포함 각 베트를 셀 (150000-200000 HSPCs 또는 100000-1000000 T 세포)의 20 µ L를 추가 합니다.
3. RNP Electroporation
- Electroporate는 nucleofector에 그들을 배치 후 큐 벳. HSPCs, 펄스 코드 ER100 사용 하 여. T 세포에 대 한 펄스 코드 EH-115를 사용 합니다.
-
HSPCs만: 보충된 SFEM 매체 (37 ° C로 예 열) electroporation 고 셀 10-15 분에 대 한 복구 후 즉시 각 베트의 추가 100 µ l
- 문화 96 잘 라운드-아래쪽에 접시와 24 h에 대 한 보충된 SFEM 매체의 추가 100 µ L을 추가 하는 셀을 전송 합니다.
- 신선한 보충된 SFEM 매체에 그들을 변경 하 고 추가 24-72 h에 대 한 그들을 품 어.
- 유전형 셀 제거 그들 게시물 electroporation 48-96 h. 5 분에 대 한 300 x g에서 셀을 회전 하 고 DNA 추출 (8.2 단계)을 시작 하기 전에 상쾌한을 제거 합니다.
-
T 세포만: 80 µ L RPMI의 문화 미디어 (필요한 경우) 다중 채널 피 펫을 사용 하 여 각 베트 또는 잘, 저수지에서 37 ° C에 미리 예 열 완료 추가.
- 15 분 동안 37 ° C에서 그들을 품 어.
- 대상 plate(s)를 적절 한 미디어, 항 체, cytokines, 등 을 추가 하 고 미리 37 ° C 배양 기에서 그들을 따뜻하게.
- (필요한 경우) 다중 채널 피 펫을 사용 하 여 라운드 하단 96 잘 접시에 우물에서 electroporated 셀의 107 µ L를 전송.
- 에 대 한 내용은 편집 결과 평가, 8 단계로 건너뜁니다.
4. 선 충 C. 준비
-
microinjection 이전 1 일: agarose 패드는 microinjection에 대 한 준비.
- 물 agarose를 추가 하 고 솔루션 끓인 뜨거운 접시에 또는 전자 레인지에 데 려 하 여 물에 3% (w/v) agarose 솔루션을 확인 합니다.
- 24 m m x 50 m m x 1.5 m m 커버 유리 슬라이드 테이블에 정렬 하 고 슬라이드에 agarose 솔루션의 작은 (~ 15 µ L) 드롭을 유리 파스퇴르 피 펫을 사용 하 여. 신속 하 게 다른 coverslip 상단에 배치 하 여 agarose 드롭 평평. 공고히 하는 coverslips 중 하나를 제거 후 agarose를 허용 합니다.
- 탁상에 coverslip agarose 코팅 얼굴 위로 두고 건조 하룻밤. 24 시간 후 agarose 패드 깨끗 하 고 마른 컨테이너에 저장 합니다.
참고:이 사용할 수 있습니다 무기한.
- Microinjection 바늘을 당겨: 필 라 멘 트와 함께 붕 규 산 유리 모 세관을 사용 하 여 (외부 직경 1.0 m m 및 내부 직경 0.58 m m), 멜로 및 화재57 및 다른 리소스58에 따라 바늘을 당겨. 바늘은 즉시 사용할 수 있습니다 또는 클레이 지원에 의해 불타는 깨끗 하 고 마른 컨테이너에 저장할 수 있습니다.
- 벌레의 유지 보수에 대 한 선 충 류 성장 미디어 (NGM) 한 천 배양 접시에 부 어 OP50 박테리아 발견 준비 (프로토콜 표준 C. 선 충 에 대 한 유지 보수 및 성장 매체에 대 한 요리법 Stiernagle59참조).
- Microinjection에 대 한 웜 무대: 12 24 h microinjection, 이전 OP50 박테리아와 새로운 NG-한 천 배지를 광고에 나온 것 L4 무대를 선택 하 고 20 ° c.에서 그들을 밤새 품 어 각 Cas9 대상/주입 혼합에 대 한 접시에 30 ~ 벌레를 선택 하십시오.
-
Microinjection의 날: 로드 RNP 솔루션 표면에 뜨는 단계 1.5에서 가져온된 microinjection 바늘.
- (일반적으로 미만 0.1 µ L 로드) 준비 microinjection 바늘으로 모 세관 피 펫에서 가져온된 모 세관 피 펫 및 백필 솔루션 단계 1.5.4에서에서 상쾌한 플라스틱.
- micromanipulator에 연결 된 microinjection 장치에 로드 된 바늘을 탑재 합니다. 250 kPa 그리고 25 kPa에 균형 압력을 주입 장치 압력을 설정 합니다.
-
날카로운 바늘 가장자리를 생성 하기 위해 로드 바늘 팁 다시 휴식. 장소는 15 m m x 15 m m x 1.5 m m 평방 coverslip는 24 m m x 50 m m x 1.5 m m coverslip 상단.
- Halocarbon 오일 700 평방 coverslip의 한 가장자리를 오버레이 합니다.
- 15 m m 평방 coverslip의 가장자리에 기름에 바늘을 위치.
- 현미경 단계와 coverslip 가이드, 슬라이드 및 바늘의 가장자리를 따라 주입 페달/버튼 동안 브러시는 손을 사용 하 여. 바늘에서 액체의 흐름을 증가, 바늘 끝을 휴식. ~ 1 거품/s 형성 바늘의 가장자리를 따라 흐름을 섞어 주사를 함으로써 최적의 흐름 속도 얻을 수 있습니다.
- Microinjection 전에 12-24 h를 선택 하는 L4 벌레는 발달 단계적된 젊은 성인 사용 하 여 주입의 하루에 확인 하십시오. 젊은 성인 벌레 OP50 박테리아 부족 한 NG-천 배지를 선택 하 고 5 분 동안 주위를 크롤 링 하도록 허용. 이 바늘 나 막 신을 최소화 하는 주입 패드에 전송 하는 박테리아의 양을 줄일 수 있습니다.
- 절 개 범위에 주입 패드/coverslip agarose를 놓습니다. 패드의 한 모서리를 따라 halocarbon 기름의 작은 트랙 누워 웜 선택을 사용 하 여.
-
기름에 코팅 웜 선택 사용 하 여, 여러 벌레 NG-한 천 배지와 오일의 트랙으로 들어올립니다. 속눈썹 또는 고양이 수염 같은 피 펫에 연결 된 잘 머리와 평행, 부드럽게 agarose 패드에 벌레를 밀어에 벌레를 놓습니다. Microinjection 절차와 함께 편안 하 게 때까지 탑재 하 고 한 번에 한 벌레를 주사.
참고: 건조 agarose 패드에 그들을 일으키는 벌레에서 수 분을 심지 것입니다. 따라서, 하나 해야 작동 신속 하 게 벌레를만.- 한 번에 놓고 몇 방울 벌레의 끝에서 halocarbon 오일 (~ 20 µ L)의 선택 또 다른 벌레를 오버레이 패드에 연결 된.
5. 선 충 C. 생식 Microinjection RNPs 및 포스트 주입 관리
참고: microinjection 프로토콜 멜로 화재57에서 적응 하 고 다른 곳에서 상세히 설명60,61.
-
주사 현미경에 탑재 된 웜 coverslip을 놓습니다. 낮은 확대 (5 X 목표, 10 배 눈), 아래 벌레 주사 바늘에 수직 위치.
- 고배율 (40 X 목표, 10 배 눈), 전환 핵에 중순-하 늦은-pachytene 근처 지역에 해당 하는 생식 선 팔에 인접 한 바늘 위치를 변경할.
- micromanipulator를 사용 하 여 바늘 웜, 표 피를 약간 우울에 대 한 이동 합니다. 그런 다음 한 손으로 표 피를 통해 바늘을 맞으면 현미경 무대의 측면을 누릅니다. 사출 페달/버튼을 눌러 천천히 주입 혼합 생식 선 팔을 채울 하 고 바늘을 제거.
- 다른 생식 선 팔이 단계를 반복 합니다.
-
일단 벌레 주입 coverslip/agarose 패드를 제거 하 고 해 현미경 아래 장소.
- 가져온된 모 세관 피 펫을 사용 하 여, 그들에 M9 버퍼를 pipetting으로 벌레에서 기름 치환. 이 치료는 agar에서 웜 해제를 수행 합니다.
- 10 분 후 때 벌레는 버퍼에서 주위 탈 곡, 들 이동 한 NG-천 배지 가져온된 모 세관 피 펫을 사용 하 여 OP50 박테리아. 벌레 회복 되며 주위에 이동 될 때까지 2-3 h 20 ° C에서 접시를 놓습니다.
- 일단, 개별적으로 OP50와 NG-한 천 배지를 벌레를 전송 및 25 ° C 인큐베이터에 접시를 전송.
-
P0를 허용-성장 하 고 3 일에 대 한 자손을 낳 기 벌레를 주입. F1 자손 화면.
- 공동-CRISPR 또는 공동 변환62,를 사용 하 여63,64,65에 참조 유전자의 돌연변이 형 포함 여부에 따라 심사에 대 한 후보 웜 선택 합니다. 개별적으로 OP50와 새로운 NG-한 천 배지를이 표시 된 벌레를 전송 하 고 20 ° c.에 F2 자손을 낳 기 위해 그들을 허용합니다
참고: 공동-CRISPR 검사 또는 선택에 사용 되는 표현 형 Cas9 편집의 성공에 대 한 초기 추정치를 제공 해야 합니다. - Co CRISPR 표현 형 없으면 microinject microinjection 효율 향상에 도움을 긍정적인 제어 플라스 미드.
참고: 예를 들어, 사출 믹스 묘-2 mCherry 태그를 인코딩하는 플라스 미드를 포함 하 여 도울 것 이다 주입 효율을 평가. 웜 성공적으로 pCFJ90 주사 형광 pharynxes와 일부 자손을 가질 것 이다.
- 공동-CRISPR 또는 공동 변환62,를 사용 하 여63,64,65에 참조 유전자의 돌연변이 형 포함 여부에 따라 심사에 대 한 후보 웜 선택 합니다. 개별적으로 OP50와 새로운 NG-한 천 배지를이 표시 된 벌레를 전송 하 고 20 ° c.에 F2 자손을 낳 기 위해 그들을 허용합니다
- 원하는 편집의 존재에 대 한 F1 벌레를 검사 합니다. 96 잘 접시의 개별 음을 F1 어머니를 선택 하 고, 그녀를 lyse 삽입 특정 PCR 증폭, DNA 순서 분석, 또는 측량 nuclease 분석 (CEL-1) 결과66에 의해 그녀의 DNA를 검사.
참고:이 분석 실험 때 공동-CRISPR/co conversion 또는 다른 검사 또는 선택 정권65,66,,6768을 사용 하 여 수행할 수 있습니다. - 에 대 한 내용은 편집 결과 평가, 8 단계로 건너뜁니다.
6. P. hawaiensis 준비
- 이전에 microinjection, 1 일 전에; 밤 '쌍 탱크'를 설정 하 여 초기 배아에 대 한 풍부 새로 분리 된 여성 갓 수정 된 배아를 포함 됩니다. Rehm 외. 참조 자세한 내용은 69 .
- Microinjection 당일 수집 단일 셀 Parhyale 배아 (0-4 h 후 수정) 바닷물에 0.02% 정 향 기름으로 벗 여성 마비와 부드럽게 뽑아 불꽃을 사용 하 여 그녀의 복 부 쪽 주머니에서 배아를 근 근이 고 둥근된 유리 피 펫 그리고 #3 집게의 둔 한 쌍.
7. P. hawaiensis 태아 Microinjection RNPs 및 포스트 주입 관리
- 백필 RNP 주입 혼합의 약 1 µ L 가져온된 모 세관 튜브 위에서 설명한.
-
Rehm 외 에 설명 된 대로 각 배아를 microinject 압축된 질소를 사용 하 여 69.
- 주사는 microinjector를 사용 하 여 한 micromanipulator 해 현미경 Parhyale 배아. 가져온된 모 세관 튜브 (4 인치-1.0 m m 사용 하 여 장치를 당기는 micropipette 뽑아 필 라 멘 트와 함께)의 뒤쪽으로 사출 믹스의 1.5 µ L 로드 microloader 피 펫 팁을 사용 하 여.
- 사출 장치에 바늘을 설정 하 고 휴식 해 범위에서 집게의 쌍을 사용 하 여 (아주 소량) 바늘의 팁. 보정 볼륨 halocarbon 오일 700 주입 거품의 직경을 측정 하 여 전달.
- 잘라 면도날을 사용 하 여 경화 에이전트에서 '구 유'. 그것은 중간 필터 멸 균 해 수로를 안정화 하 여 물에서 Parhyale 배아를 일렬로.
- 주사 주입 하는 동안 집게의 쌍을 가진 각 배아를 안정화 microinjection 설정을 사용 하 여 배아. 주입 후 유리 전송 피 펫을 사용 하 여 중간 필터 멸 균 해 수 가득 신선한 60mm 문화 접시 배아 전송.
-
1 부는 형태는 2-셀 배아 (4-6 h 후 수정) 이미 발생 한 경우 두 blastomeres 주입 하 여 완전히-돌연변이 동물을 생성 합니다. 2 셀 무대의 총 절단 되도록 공동 blastomeres는 FITC 또는 TRITC dextran 주입 하 고 신호는 단일 blastomere 형광 사출 후 범위 해 부의 밑에 제한 관찰 합니다.
- 또는, 2 세포 단계 (조직 및 A P 축 위치에 따라 대략 분할된 왼쪽-오른쪽)에서 두 blastomeres 중 하나만 주입 하 여 ' 반-돌연변이 ' 동물을 생성 합니다.
- 8 세포 배아 (7.5-9 h 후 수정) 세균 층에 편집을 제한 하에 한 셀을 주입. Gerberding 외. 참조 70 초기 blastomere 계보의 지도
-
60mm 문화 요리 (더 이상 접시 당 25), '전 산소' 수족관 bubbler를 사용 하 여, 필터 멸 균 해 수를 사용 하거나 적극적으로 떨고 중간 가득에 태아를 품 어.
- 습도 유지 하 여 12 h 빛 어두운 주기와 26 ° C 배양 기에 넣어 젖은 종이 타월로 줄지어 느슨하게 봉인 plasticware에 배아의 요리를 놓습니다.
- 청소 해 요리 마다 몇 일 살아남은 배아를 전송 합니다.
참고: 배아 실내 온도에, 비록 그들은 훨씬 더 느리게 개발 양식 수 있습니다.
-
해 부와 제자리에서 교 잡 또는 항 체 얼룩 (참조 브라운 외. 여 식 분석에 대 한 다양 한 단계에서 배아를 해결 71 준비 가이드, 및 해 부 및 고정72, 현장에서 교 잡73,74를 얼룩이 지는 항 체에 대 한 추가 참조).
- 약 텅스텐 와이어의 구부러진된 부분을 스레딩 하 여 절 개 바늘을 만들기는 인슐린 바늘의 끝에 길이 0.5. 전류에서 수산화 나트륨에에서 바늘을 선명 하 게. 1 mL 주사기를 사용 하 여 절 개 바늘의 손잡이로.
- 9 부품 PEM 버퍼 (파이프 pH 6.95, EGTA, 2 mM MgSO4의 1 m m의 0.1 m M), 1-부분 10 x PBS, 및 1 개 부품 32%의 갓 만든 솔루션 3-잘 유리 접시 중간의 한 잘 채워 PFA. 3-5 배아를 접시에 놓고 구멍 작은 안정, 구멍을 약간 무디게 한 날카로운 텅스텐 바늘을 사용 하 여 각 태아에 밖으로 흐르는 노른자위와에서 실행 되도록 정착 액.
- 날카롭게 텅스텐 바늘의 한 쌍을 사용 하 여, 부드럽게 놀 릴 거리 Parhyale 태아를 둘러싼 외부 두 막. 더 강력한 배아를 위해 정착 액에 그들을 해 부만 막 막 제거를 더 어렵게 만드는 배아에 고정 되 고 하는 것을 막기 위해 신속 하 게 작업. 항 체 얼룩이 지기에 대 한 15-20 분 또는 40-50 분에 제자리 교 잡에 대 한 총 수정 배아를 허용 합니다.
- 이미지 라이브 파묻혀 고 형태학과 행동 고기에 대 한 분석 또는 해결 하 고 더 자세한 분석을 위해 그들을 얼룩. 성적 성숙에 파묻혀를 녹아웃과 유전자 변형 라인 (Kontarakis 및 Pavlopoulos75 hatchling 관리 및 다른 유용한 정보에 대 한 참조)을 2-3 개월에 올립니다.
8. 평가 결과 편집
- 해당 되는 경우 시각적 또는 기능 형 편집된 셀 또는 유기 체에서 찾습니다.
참고:이 프로세스 달라 집니다 널리 응용 프로그램, 그리고 몇 가지 예제 위의 그들의 관련 프로토콜 단계의 끝에 설명 되어 있습니다. HSPCs 낫 세포 돌연변이 수정 후 헤모글로빈 생산 차별화 된 erythroblasts HPLC (그림 1A)를 사용 하 여 분석. T 세포에 일리노이-2 수용 체 유전자의 녹아웃과 표면 얼룩에 의해 확인 될 수 있다 cytometry (그림 1B) 합니다. C. 선 충 및 P. hawaiensis 고기 평가, 동물 형태학 및 빛 또는 형광 현미경 (그림 1C 및 1d) 동작을 관찰 합니다. - 효율성과 생성 된 게놈 편집의 종류를 확인 하려면 편집된 셀의 풀을 lyse 하 고 상업 추출 키트21을 사용 하 여 그들의 게놈 DNA를 추출 합니다.
-
Indel 형성의 빠른 추정 컷 주위 자료 쌍 PCR 증폭 적어도 200 사이트 및 수행을 T7 endonuclease1 (T7E1)76 또는 측량 (CEL-1 nuclease) 분석 결과77.
- Cas9 컷 사이트 또는 성공적인 HDR indel 형성 됩니다 만들거나 알려진된 제한 사이트를 제거 하는 경우 편집 효율성6추정 하는 금지 효소 소화를 사용 하 여 하는 것이 좋습니다. 금지 파편 길이 다형성 (RFLP) 분석 결과 그것을 사용할 수 경우 효율성을 확인 하기 편리한 방법일 수 있다.
- 표준 생어에 대 한 PCR amplicon 보낼 편집 효율의 정확한 정량화 및 주된 편집 결과의 결정에 대 한 정방향 및 역방향 뇌관으로 시퀀싱.
참고: 단일 클론 또는 유기 체를 분석 하는 경우 생어 결과의 분석은 간단 하 고, 그림 2A에서 같이. 셀의 풀을 분석 하는 경우 다음 그림 2B와 같이 온라인 도구78와 chromatograms 분석. - 그림 2C에서처럼 전체 정량화 및 결과 편집의 시퀀스에 대 한 깊은 시퀀싱27,54을 수행 합니다.
- 오프 대상 변경, PCR 증폭 예측된에서 대상 사이트의 특정 집합을 평가 하려면 NGS 위해 보내. 염색체 translocations의 검색을 사용 하려면 가이드-seq79 또는 높은 처리량, 게놈 넓은 전 시퀀싱 (HTGTS)80을 수행 합니다. 클론 인구에서 오프 대상 편집의 완전 한 그림에 대 한 전체 게놈 시퀀싱 (WGS)81,,8283을 수행 합니다.
참고: 설명에-및 오프-대상 게놈 편집 측정 더 다양 한 검토에 기사84,,8586에 대 한 방법의 다양 한이 있습니다.
Representative Results
이러한 실험 쇼 어떻게 조립된 Cas9 RNP 기본 세포와 모든 생물의 유전자 조작에 사용할 수 있습니다. 연구원 정화 또는 구매 Cas9 단백질 및 sgRNA, 콤플렉스를 미리 구성 하는 두 요소를 결합 하 고 그들의 세포 또는 관심의 유기 체에는 RNP 소개. 후 발생 및 출생 (있는 경우) 다음 세대의 자손, 고기에 대 한 확인 및 수집 유전형 셀 편집을 위한 충분 한 시간을 허용 합니다. 고기는 기능적인 분석 실험, 식 분석, 시각화 (눈 또는 현미경 검사 법), 또는 실험에 따라 다른 방법을 통해 관찰 될 수 있습니다.
예를 들어 HSPCs 낫 세포 질병의 원인 β-globin 돌연변이 해결 하기 위해 편집 된 적혈구로 분화 수 하 고 건강 한의 생산 또는 겸 상 적혈구 헤모글로빈27,87 (그림 1에 대 한 분석 A). T 세포는 높은 선호도 일리노이-2 수용 체 유전자, CD25 밖으로 노크 편집 (IL2RA), 수 표면 얼룩 및 교류 cytometry88, 분석 하 고 일리노이-2 자극 (그림 1B신호 응답을 감지 하기 기능 분석 ). T 세포는 HIV 감염89 의 효능을 포함 하 여 다른 고기에 대 한 평가 필요로 하는 많은 임상으로 중요 한 방법으로 재설정 또한 수 그리고 vivo에서 antitumor 효능 자동차 T의 세포11.
공동-CRISPR/co conversion 심사 방식을 사용 하 여, C. 선 충 벌레 두 loci62에서 동시에 편집 됩니다. Dpy 10 참조 유전자는 ssODN를 사용 하 여에 HDR 쉽게 득점 지배적인 dpy 10 이득의 기능 돌연변이에서 템플릿 결과 복구 합니다. Heterozygous F1dpy-10(gof) 동물 롤러 (Rol) 이며 homozygous dpy-10(gof) 동물은 땅딸막한 (Dpy). 표현 형의 존재는 Cas9 편집이 동물에서 발생 나타내고 식별 Rol 또는 Dpy F1 동물에서 두 번째 장소에서 편집 이벤트의 확률을 향상 시킵니다. 성공적인 편집 실험 주입된 P0 웜 저조한 Rol 또는 Dpy90는 20 이상의 F1 자손의 33-50% 발생 한다. 그것은 다음 wildtype dpy 10 반환 하 여 관심의 homozygous 편집에 대 한 선택 비 Rol 동물을 선택 수 있습니다. 엄지손가락의 규칙으로 서 co CRISPR 참조 유전자를 대상으로 하는 crRNA의 농도 대상으로 관심사의 유전자는 crRNA의 그 절반 이어야 한다. 관심사의 유전자에는 편집, 복구 되지 않은 경우 원하는 돌연변이 복구의 가능성을 높이기 위해 두 CRISPR RNAs의 비율을 조정할 수 있습니다. 예를 들어, 참조 유전자 crRNA에 상대적인 관심사의 유전자에 대 한 crRNA의 양을 증가 하는 것은 벌레 벌레는 또한 참조 유전자 소재 시에 편집의 인구 내의 관심사의 유전자에서 편집 보유의 비율을 증가 합니다. 공동 변환 주파수가 다르지만 요금은 일반적으로 20-60%, F1 세대 (그림 1C)에서 homozygous 편집 자주 떨어졌다.
P. hawaiensis 파묻혀 복 B 유전자 (Abd-B) 밖으로 노크 편집 표시 분명 형태학 이상이3 (그림 1D). 이 유전자는 올바른 복 부 패턴화 하기 위한 필요 하며 흉부 유형 점프 및 교체는 일반적으로 수영, 앵커 다리 다리 걷기의 중단 결과 복 부에 존재.
결정 하는 genotypic 수준에서 결과 편집 하는 게놈 시퀀싱 또는 생체 외에서 분석 결과 시퀀스 변경 사항을 감지 하는 필요 합니다. 여기, 우리가 표시 대표 시퀀싱 데이터에서 우리의 모델 세포 유형 및 유기 체, 정량화 편집에 다른 접근을 강조. 그림 레이블 모든 방법을 여기에 표시 된 어떤 생물 학적 시스템에 적용 될 수 있기 때문에 일반화는 note.
시퀀싱-기반 접근 기술적인 복잡성과 결과의 깊이에서 변화 한다. 클론 편집된 인구 또는 쉽게 분리할 수 있는 개별 유기 체, 편집된 개인 게놈 DNA 추출 다음 시퀀싱 수 있습니다. 표준 생어 시퀀싱 결과 순서 변경 기능 (그림 2A)를 방해 것이 가상 frameshifts와 주어진된 개인에서 Cas9 컷 사이트에서 발표할 예정 이다. 시퀀싱에 사용 되는 온라인 도구가입니다 다른 생어 시퀀싱 기반 접근 방식을 개별 돌연변이78보다는 오히려 혼합된 인구에 적용 될 수 있는. 시퀀스는 주된 시퀀스 결과 뿐 아니라 전체 편집 효율 대략적인 수 있는 온라인 도구와 분석 된다. 대표적인 데이터 그림 2B에 나와 있습니다.
여기 설명 하는 가장 철저 한 시퀀싱 방법을 깊은 시퀀싱 (높은 처리량 또는 다음-세대 시퀀싱 라고도 함)입니다. 이 메서드는 혼합된 인구에 개별 게놈에서 DNA 시퀀스를 제공합니다. 이러한 데이터 다양 한 방법으로 설명 될 수 있다. 여기, 우리 편집 결과 (그림 2C)에 따라 편집된 셀에서 개별 연속 읽기를 분류 했습니다. 대부분의 세포는 일반적으로 유전자 장애 귀착되는 NHEJ 통로 통해 편집 됩니다. 다른 사람에서는, HDR27를 통해 다른 버전에 대 한 대상 유전자 스와핑할 되어 있다.
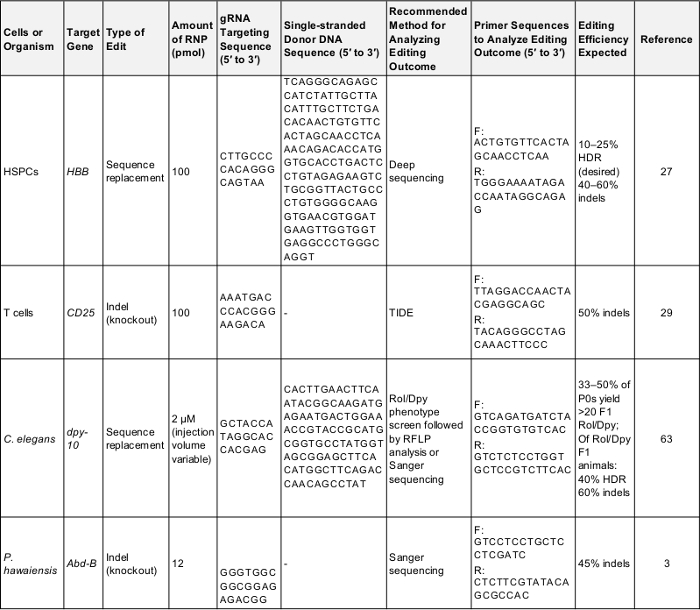
표 1: 긍정적인 예비 게놈 실험 편집에 대 한 제어. 이 표에서 처음 게놈 편집 하는 각 셀이이 프로토콜에서 설명 하는 유기 체에서 실험을 수행 하는 데 필요한 중요 한 정보를 보여 줍니다. 이러한 매개 변수를 다음 프로토콜을 테스트 하는 데 사용할 수 있는 성공적인 결과가 것 또는 비교를 한 번에 대 한 기준으로는 실험 대상으로 그들의 자신의 관심사의 유전자. F: 앞으로, r: 리버스, HDR: homology 감독 수리. 이 테이블을 다운로드 하려면 여기를 클릭 하십시오.

그림 1 : 대표 phenotypic 결과 Cas9 RNP 편집 기본 인간 세포와 유기 체에서. (A) 후 성공적인 게놈 편집, HSPCs 단계의 erythroblasts로 분화 하는 겸 상 적혈구 헤모글로빈 보다는 더 많은 기능 헤모글로빈을 생산할 예정 이다 보여주는 HPLC 추적입니다. 돌연변이 적혈구 생산 겸 상 적혈구 헤모글로빈 (HbS) 동안 성공적으로 편집 셀 태아 헤모글로빈 (HbF) 뿐만 아니라 건강 한 헤모글로빈 (HbA와 HbA2)을 생산할 예정 이다. 흡수도 임의 단위 (au) 그래프. 이 패널 처음 윗 et 그리고 다른 곳 에 게시 된 27. 과학의 전진을 위한 미국 협회에서 허가로 증 쇄 하는. (B) 각 조건에 대 한 왼쪽에이 패널 CD25 유전자 RNP와 제압 된 후 T 세포 표면 얼룩이 CD25를 표현 하지 않습니다 보여주는 흐름 cytometry 데이터를 보여줍니다. CD25 풍부 x 축에서 y 축에 셀 크기와 플롯 됩니다. 각 조건에 대해 오른쪽이 패널 일리노이-2와 유도 후 인-Stat5 (pStat5) 부 량을 보여줍니다. 신호는 일리노이-2 수용 체 (CD25 코) 결 석 때 감소 된다. PStat5 풍부는 x 축에 표시 하 고 일리노이-2 입력의 세 가지 다른 수준에서 발생 하는 데이터는 수직으로 비교 됩니다. (C)이 이 패널 꼬마 선 충 공동-CRISPR/co conversion 화면 dpy 10 공동 변환 표식으로 타겟팅을 보여줍니다. 2 안내 RNAs 대상 두 loci, dpy-10 당신의 좋아하는 유전자 (yfg), 동일한 P0에서-주입 된 동물. Dpy 10 에서 HDR 결과 Rol 또는 Dpy 표현 형. Rol 또는 Dpy F1 동물의 선택 하면 식별 하는 두 번째 장소에서 편집의 가능성이 높아집니다. (D)이 이 패널 wildtype Parhyale hawaiensis 파묻혀 수영 및 앵커 다리 정상적인 인체를가지고 있음을 보여준다. Abd-B 노크 아웃 파묻혀 (F0 개인)는 복 부 흉부 쪽으로 변형 개발. 따라서, 수영 및 앵커 다리 간은 고 정상 흉부와 관련 된 점프 그리고 다리에 의해 대체. 이 그림의 더 큰 버전을 보려면 여기를 클릭 하십시오.

그림 2 : 편집 결과 분석 방법에서 전형적인 결과. (A) 가이 패널에서 개별 F1 P. hawaiensis , 포함 한 유기 체 wildtype 시퀀스 및 열려있는 독서 프레임을 이동 하 여 유전자 기능을 방해 하는 3 개의 다른 indels 시퀀싱 결과 생어의 예를 보여줍니다. (B) 이 조 수 결과 삽입 및 시퀀스 된 T 세포의 수영장에서 Cas9 대상 사이트에서 발생 한 삭제 이벤트의 범위를 보여. X 축 지정된 삽입 또는 삭제 뉴클레오티드에의 길이 나타냅니다. (C) 이러한 깊은 시퀀싱 결과 표시 없이 nucleofection 또는 gRNA, 아무 게놈 편집 및 성공적인 그대로 Cas9 RNP와 편집, 결과별 DNA 수리 HSPCs. 이 그림의 더 큰 버전을 보려면 여기를 클릭 하십시오.
Discussion
강력한 게놈 프로토콜 편집 설정 셀에 선 또는 관심의 유기 체 요구 최적화 하 고 경험적이 섹션에서 설명한 몇 가지 주요 매개 변수 테스트. 여기에 제시 된 일반적인 접근 방법의 몇 가지 유사 콘텐츠를 시도 하는 것은 매우 좋습니다. 이 프로토콜의 주요 한계는 다른 셀을이 메서드를 적용 하는 또는 생물 공부, 수 종에 따라 다른 결과가 발생할 수 있습니다 및 높은 효율 유전자 녹아웃 하는 실험적인 디자인 DNA 삽입을 홍보 하지 않을 수 있습니다. 따라서, 여기에 제시 된 및 아래에 설명 된 문제 해결 방법으로 시작 하는 것이 좋습니다.
게놈 편집 시 품질 문제 해결:
생성 또는 높은-품질의 시 약을 구입 어떤 게놈 프로토콜 편집에서 중요 한 단계입니다. Cas9 단백질 실험실에서 정화 하거나 상업적으로 구매 될 수 있습니다. 많은 프로토콜 RNP 조리법, Cas9에 대 한 최종 농도 참고 하지만 최적의 유전자 활동 편집 소스에 따라 달라 집니다 어떤 개별 Cas9 단백질 준비의 구체적인 활동에 따라 달라 집니다. 여기에 제시 된 프로토콜 동작 고려 RNP 넣는 Cas9 수준에 의해 최적 농도 설정 하는 데 사용의 양을 최적화: 불필요 한 대상에서 분열으로 인 없이 매우 구체적인 대상 DNA 분열을 제공 과도 한 Cas940.
가이드 RNA의 순도 균질 성공22편집 게놈의 결정을 수도 있습니다. 구입한 sgRNAs 또는 별도 crRNA 및 tracrRNA 구성 요소는 일반적으로 높은 품질의 시 약 하 고 다양 한 화학 수정 RNA 저하 문제를 방지 하거나 스며들 게 RNP91에 추가 기능을 사용할 수 있습니다. 화학적 수정 gRNAs 표준 게놈 편집 실험에 필요한 되지 않을 수 있습니다, 일부 단체는 훨씬 더 높은 효율성과 같은 시 약, 그래서 그들은 과정을 습득 후 시도할 만한 가치가 있을 수 있습니다 또는 때 편집 관찰 gRNA 저하 문제22,91나타납니다. 생체 외에서 전사 및 후속 젤 정화 실험17,21,,4950편집 일상적인 게놈에 대 한 충분 한 될 수 있는 저렴 한 대안 이다. 또한, 일반적으로 균질 성 gRNA 인구에서 vivo에서, 개별 가이드의 절단 ribozyme 및 tRNA 기반을 포함 한 생산에 적용, 연장 될 수 있습니다 체 외에서 RNA 준비 청소기를 생성 하는 몇 가지 방법을 제품92
가이드 RNA와 DNA 기증자 디자인 팁:
가이드 RNA 선택 대상에서 분열의 가능성을 최소화 하면서 매우 효율적인 대상에 편집 달성에 중요 한 요소 이다. 가이드 선택에 도움이, 몇몇 연구는 컴파일하는 데 사용 높은 처리량 화면 다음-세대 시퀀싱과 함께 성공적인 가이드47,79,,9394의 시퀀스 기능 , 9596. 이러한 기능은 예측 알고리즘 및 가이드 선택44,45,,4647,48에 도움을 온라인 도구를 개발 하기 위해 사용 되었습니다. 이러한 알고리즘은 가이드 RNA 식에 대 한 DNA 기반 시스템을 사용 하 여 화면에 기초 하 고. 가이드는 Pol III 발기인을 사용 하 여 표현 되 고 그들의 표정은 따라서 조기 종료 때 마주치의 uracil97,98, 트랙 등 유류 III 전사와 관련 된 제한 하는 경향이 99. 그러나, RNPs 사용 하 여 만든 체 외에-종합된 가이드 RNAs 그 우려를 무시 하 고 가이드 디자인에 제약을 간소화. 이 알고리즘에서 등장 하 고 매우 효과적인 게놈 편집과, 다 수의 연구에서 확인 되었다 일반적인 기능 퓨 린, 특히는 구 아닌, 가이드의 특정 대상 시퀀스의 3 ' 끝의 존재 이다. 이 가이드 기능 C. 선 충, 초파리, 그리고 zebrafish65,,100101포유류에서 배열 하는 생물 중 매우 성공 했다. 또한, C. 선 충에 대 한 가이드의 대상 지역의 3 ' 끝에 GG 디뉴클레오티드 가이드 디자인은 매우 효과적인 가이드 RNAs65예측을 위한 효과적인 전략. 이상적으로,는 특정된 응용 프로그램에 대 한 가장 성공적인 결정 하는 동시에 여러 명의 가이드를 테스트 합니다.
DNA 순서는 게놈으로 소개 하려고, 할 때 기증자 또는 템플릿 DNA의 디자인은 또한 중요 한. 단일 가닥 oligonucleotide 기증자 (ssODNs) 다른 일반적인 복구 템플릿, 선형 더블-좌초 및 플라스 미드 DNA54,,55102보다 더 안정적으로 삽입 됩니다. 일부 loci에서 비 대상에 보완 또는 DNA 물가 전치 고 상 동 팔 길이27,55에 비대칭을 ssODNs와 HDR 효율성을 개선할 수 있습니다. 복구 템플릿 컷된 사이트에 삽입 되 고 있기 때문에 대상된 시퀀스를 포함, 기증자 DNA 게놈 삽입 전후 고착에서 Cas9를 방지 하기 위해 단계를가지고 한다. 이 하 침묵 돌연변이 팸 시퀀스 또는 씨앗 지역 삽입된 유전자21,103의 기능을 유지 하면서 Cas9에 의해 인정을 피하 여 수행 됩니다. PAM에도 단일 뉴클레오티드 변화 바인딩104폐지 가능성이 있지만, 안전을 위해 적어도 4 개의 뉴클레오티드를 변경 하려고 합니다.
의미와 미래의 응용 프로그램:
CRISPR Cas9와 함께 편집 하는 게놈 어떤 유기 체의 손쉬운 유전자 조작 사용 강력한 방법으로 떠오르고 있다. 편집 Cas9 RNP와 처음에 조금 더 많은 노력을 소요 아니지만 나면 시 약 및 프로토콜은 실험실에서 사용 하는 것은 간단. 적은 대상 오프 효과24,,2526 HDR를 통해 달성 하기 어려운 유전자 삽입을 포함 하 여 더 높은 전반적인 편집 효율성 리드 플라스 미드 DNA 대신 조립된 RNP와 셀 편집 , 27 , 29. 경험와 유전자 발현, RNA 저하, 단백질 폴딩, gRNA 및 Cas9 분자 셀22,23내 별도로 합성 간의 연결 문제를 방지 하는 더. 또한 insertional mutagenesis에 대 한 안전 우려를 circumvents RNP 편집 및 바이러스 전달 방법 됩니다 때 발생할 수 있는 지속적인된 식14임상 사용. 이러한 이점 때문에 전 임상을 실시 하는 많은 과학자 개념 증명 실험 부탁 RNP 인간의 치료 응용 프로그램에 대 한 편집. 둘 다 vivo에서 및 전 비보 RNP 기반 게놈 편집 접근을 하거나 심지어 조건, 듀 켄 씨 근이 영양 증105 및 낫 세포 질병27 같은 유전 질환에서의 다양 한 개발은 에이즈29 고 암11. 흥미롭게도, Cas9 RNP는 점점 농업 공학 식물33,,3436의 편집 ' DNA 무료 ' 수 있기 때문에 대 한 전달 방법으로 사용 됩니다.
Disclosures
저자 알렉산더 Marson와 야 곱 E. 옥수수는 스포트라이트 치료제의 공동 설립자. 야 곱 E. 옥수수 임무 치료제 고문 이며 그의 실험실에서 아 스 트 라와 화 이자 후원된 연구 지원을 받고 있다. 알렉산더 Marson는 주노 치료제와 협정 치료제, 고문 그리고 그의 실험실 주노 치료제, Epinomics, 및 사노 피에서 후 원하는 연구 지원을 받고 있다. 그의 실험실은 또한 Cas9 RNP 기술 관련 특허에 대 한 적용 됩니다.
Acknowledgments
우리는 우리의 실험실과 이러한 방법의 개발에 그들의 공헌에 대 한 베이 지역 게놈 편집 커뮤니티의 많은 이전 일원을 감사합니다. 우리는 비판적으로이 원고를 읽고에 대 한 로스 윌슨을 감사 합니다.
제이크 Aronov 선물 알렉산더 Marson의 연구를 지원 하 고 국가 다 발성 경화 증 사회 (CA 1074-A-21)을 부여 합니다. 알렉산더 Marson 버로우즈 Wellcome 기금에서 의료 과학자에 대 한 경력 수상을 보유 하 고 찬 주 커 버그 Biohub 탐정입니다. 야 곱 E. 옥수수의 연구는 재생 의학에 대 한 리 카 싱 재단, 유산 의료 의료 연구소, 그리고 캘리포니아 연구소에 의해 지원 됩니다. Behnom Farboud와 바바라 J. 마이어의 연구 부분에 NIGMS 보조금 R01 GM030702 바바라 제이 메이어, 누가 하 워드 휴즈 의학 연구소의 조사에 의해 자금입니다. 에 린 자비스와 Nipam 헤 파 텔의 연구는 일부 NSF 그랜트 IOS-1257379에 의해와 린 자비스 인정 NSF GRFP와 Philomathia 대학원 장학금 지원.
Materials
| Name | Company | Catalog Number | Comments |
| Reagents/Materials | |||
| DNA oligonucleotides | Integrated DNA Technologies | - | IDT will provide custom DNA sequences, including those in Table 1 |
| Guide RNAs | Synthego | - | Synthego will provide high-quality sgRNAs for S. pyogenes Cas9, including custom sgRNAs containing the targeting sequences included in Table 1 |
| Purified Cas9 protein (EnGen Cas9 NLS, S. pyogenes) | New England Biosciences | M0646T | If possible, purifying Cas9 in-house or purchasing from local core facilities is a less expensive option |
| Normal peripheral blood CD34+ stem/progenitor cells | AllCells | PB032-2 | |
| StemSpan SFEM | StemCell Technologies | 09650 | |
| StemSpan CC110 | StemCell Technologies | 02697 | |
| P3 Primary Cell 4D-Nucleofector X Kit | Lonza | V4XP-3032 | |
| RPMI-1640 Medium, With sodium bicarbonate, without L-glutamine, liquid | Sigma | R0883-6X500ML | |
| EasySep™ Human T Cell Isolation Kit | Stemcell | 17951 | |
| cell culture plate, 96 wells, round | Fisher Scientific | 3799 | |
| CTS (Cell Therapy Systems) Dynabeads CD3/CD28 | Life Tech | 40203D | |
| Reombinant Human IL-2 | UCSF Pharmacy | NA | |
| SepMate-50 500-pack IVD | Stemcell Technologies | 85460 | |
| OP50 Escherichia coli | Caenorhabditis Genetics Center | OP-50 | https://cgc.umn.edu/ |
| Nematode Growth Media agar in petri dishes | - | - | See Stiernagle, T (ref. 59) |
| Standard borosilicate glass capillaries with filament: 4 in (100 mm), 1/0.58 OD/ID | World Precision Instruments | 1B100F-4 | |
| Single-barrel standard borosilicate glass capillaries: 6 in (152 mm), 2/1.12 OD/ID | World Precision Instruments | 1B200-6 | |
| Cover glass; 24 × 50 mm | Thermo Fisher Scientific | 12-544E | |
| Cover glass; 22 × 22 mm | Thermo Fisher Scientific | 12-518-105K | |
| Apex LE agarose | Genesee Scientific | 20-102 | |
| Halocarbon oil 700 | Sigma-Aldrich | H8898-100ML | |
| pCFJ90 plasmid | Addgene | 19327 | |
| Compressed nitrogen | - | ||
| 60 mM culture dishes | BD | ||
| Capillary tubes with filament: 4 in (1.0 mm) | World Precision Instruments | T2100F-4 | |
| Sylgard 184 | Dow Corning | ||
| Petri dishes (100 × 15 mm) | - | ||
| Tungsten wire (0.005 in. diameter) | Ted Pella | ||
| Perfluoroalkoxy alkane (PFA) | - | ||
| Marine salt | - | ||
| 9" pasteur pipettes | - | ||
| Phenol red | - | ||
| Nuclease-free water | - | ||
| Equipment | |||
| 4D Nucleofector | Lonza | AAF-1002X | |
| MZ75 Stereomicroscope | Leica | Out-of-production. Current model is the M80 Stereomicroscope | |
| Axio Vert35 inverted phase contrast fluorescent microscope | Zeiss | Out-of-production. Current model is the Axio VertA.1 | |
| Laser-based micropipette puller (for C. elegans protocol) | Sutter Instrument | FG-P2000 | |
| Picoliter Microinjector (for C. elegans protocol) | Warner Instruments | PLI-100A | |
| Three-axis Joystick oil hydraulic micromanipulator | Narishige International | MO-202U | |
| Coarse manipulator | Narishige International | MMN-1 | |
| Micropipette puller (for P. hawaiensis protocol) | Sutter Instrument | P-80/PC | |
| Microinjector (for P. hawaiensis protocol) | Narishige | IM300 | |
| Microloader pipette tips | Eppendorf | 5242956003 | |
| NG-agar |
References
- Komor, A. C., Badran, A. H., Liu, D. R. CRISPR-based technologies for the manipulation of eukaryotic genomes. Cell. , 1-17 (2016).
- Barrangou, R., Horvath, P. A decade of discovery: CRISPR functions and applications. Nature Microbiology. 2, 1-9 (2017).
- Martin, A., Serano, J. M., et al. CRISPR/Cas9 mutagenesis reveals versatile roles of Hox genes in crustacean limb specification and evolution. Current Biology. 26 (1), 14-26 (2016).
- Goldstein, B., King, N. The future of cell biology: emerging model organisms. Trends in Cell Biology. 26 (11), 818-824 (2016).
- Mali, P., Yang, L., et al. RNA-guided human genome engineering via Cas9. Science. 339 (6121), 823-826 (2013).
- Cong, L., Ran, F. A., et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 339 (6121), 819-823 (2013).
- Deltcheva, E., Chylinski, K., et al. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature. 471 (7340), 602-607 (2011).
- Jinek, M., Chylinski, K., Fonfara, I., Hauer, M., Doudna, J. A., Charpentier, E. A Programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 337 (6096), 816-821 (2012).
- Nowak, C. M., Lawson, S., Zerez, M., Bleris, L. Guide RNA engineering for versatile Cas9 functionality. Nucleic Acids Research. 44 (20), 9555-9564 (2016).
- Jiang, W., Cox, D., Zhang, F., Bikard, D., Marraffini, L. A. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nature Biotechnology. , 1-9 (2013).
- Rupp, L. J., Schumann, K., et al. CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Scientific Reports. 7 (1), 737 (2017).
- Graham, D. B., Root, D. E. Resources for the design of CRISPR gene editing experiments. Genome Biology. 16, 260 (2015).
- Wang, H., La Russa, M., Qi, L. S. CRISPR/Cas9 in genome editing and beyond. Annual Review of Biochemistry. 85, 2270 (2016).
- Nelson, C. E., Gersbach, C. A. Engineering delivery vehicles for genome editing. Annual Review of Chemical and Biomolecular Engineering. 7, 637-662 (2016).
- Yin, H., Kauffman, K. J., Anderson, D. G. Delivery technologies for genome editing. Nature Reviews Drug Discovery. 16 (6), 387-399 (2017).
- Zuris, J. A., Thompson, D. B., et al. Cationic lipid-mediated delivery of proteins enables efficient protein-based genome editing in vitro and in vivo. Nature Biotechnology. 33 (1), 73-80 (2015).
- Cho, S. W., Lee, J., Carroll, D., Kim, J. -S., Lee, J. Heritable gene knockout in Caenorhabditis elegans by direct injection of Cas9-sgRNA ribonucleoproteins. Genetics. 195 (3), 1177-1180 (2013).
- Wang, W., Kutny, P. M., et al. Delivery of Cas9 protein into mouse zygotes through a series of electroporation dramatically increases the efficiency of model creation. Journal of Genetics and Genomics. 43 (5), 319-327 (2016).
- Chen, S., Lee, B., Lee, A. Y. -F., Modzelewski, A. J., He, L. Highly efficient mouse genome editing by CRISPR ribonucleoprotein electroporation of zygotes. Journal of Biological Chemistry. 291 (28), 14457-14467 (2016).
- Remy, S., Chenouard, V., et al. Generation of gene-edited rats by delivery of CRISPR/Cas9 protein and donor DNA into intact zygotes using electroporation. Scientific Reports. 7 (1), 16554 (2017).
- DeWitt, M. A., Corn, J. E., Carroll, D. Genome editing via delivery of Cas9 ribonucleoprotein. Methods. , 1-7 (2017).
- Hendel, A., Bak, R. O., et al. Chemically modified guide RNAs enhance CRISPR-Cas genome editing in human primary cells. Nature Biotechnology. 33 (9), 985-989 (2015).
- Thyme, S. B., Akhmetova, L., Montague, T. G., Valen, E., Schier, A. F. Internal guide RNA interactions interfere with Cas9-mediated cleavage. Nature Communications. 7, 11750 (2016).
- Kim, S., Kim, D., Cho, S. W., Kim, J., Kim, J. -S. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Research. 24 (6), 1012-1019 (2014).
- Lin, S., Staahl, B. T., Alla, R. K., Doudna, J. A. Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. eLife. 3, 04766 (2014).
- Liang, X., Potter, J., et al. Rapid and highly efficient mammalian cell engineering via Cas9 protein transfection. Journal of Biotechnology. 208, 44-53 (2015).
- DeWitt, M. A., Magis, W., Bray, N. L., Wang, T. Selection-free genome editing of the sickle mutation in human adult hematopoietic stem/progenitor cells. Science Translational Medicine. 8 (360), (2016).
- Ramakrishna, S., Kwaku Dad, A. -B., Beloor, J., Gopalappa, R., Lee, S. -K., Kim, H. Gene disruption by cell-penetrating peptide-mediated delivery of Cas9 protein and guide RNA. Genome Research. 24 (6), 1020-1027 (2014).
- Schumann, K., Lin, S., et al. Generation of knock-in primary human T cells using Cas9 ribonucleoproteins. Proceedings of the National Academy of Sciences of the United States of America. 112 (33), 10437-10442 (2015).
- Lee, J. -S., Kwak, S. -J., et al. RNA-guided genome editing in Drosophila with the purified Cas9 protein. G3: Genes, Genomes, Genetics (Bethesda, MD). 4 (7), 1291-1295 (2014).
- Sung, Y. H., Kim, J. M., et al. Highly efficient gene knockout in mice and zebrafish with RNA-guided endonucleases. Genome Research. 24 (1), 125-131 (2014).
- Menoret, S., De Cian, A., et al. Homology-directed repair in rodent zygotes using Cas9 and TALEN engineered proteins. Scientific Reports. 5, 14410 (2015).
- Woo, J. W., Kim, J., et al. DNA-free genome editing in plants with preassembled CRISPR-Cas9 ribonucleoproteins. Nature Biotechnology. 33 (11), 1162-1164 (2015).
- Malnoy, M., Viola, R., et al. DNA-free genetically edited grapevine and apple protoplast using CRISPR/Cas9 ribonucleoproteins. Frontiers in Plant Science. 7, 1904 (2016).
- Svitashev, S., Schwartz, C., Lenderts, B., Young, J. K., Mark Cigan, A. Genome editing in maize directed by CRISPR-Cas9 ribonucleoprotein complexes. Nature Communications. 7, 13274 (2016).
- Liang, Z., Chen, K., et al. Efficient DNA-free genome editing of bread wheat using CRISPR/Cas9 ribonucleoprotein complexes. Nature Communications. 8, 14261 (2017).
- Shin, S. -E., Lim, J. -M., et al. CRISPR/Cas9-induced knockout and knock-in mutations in Chlamydomonas reinhardtii. Scientific Reports. 6, 27810 (2016).
- Pohl, C., Kiel, J. A. K. W., Driessen, A. J. M., Bovenberg, R. A. L., Nygård, Y. CRISPR/Cas9 based genome editing of Penicillium chrysogenum. ACS Synthetic Biology. 5 (7), 754-764 (2016).
- Grahl, N., Demers, E. G., Crocker, A. W., Hogan, D. A. Use of RNA-protein complexes for genome editing in non-albicans Candida species. mSphere. 2 (3), (2017).
- Rivera-Torres, N., Kmiec, E. B. A standard methodology to examine on-site mutagenicity as a function of point mutation repair catalyzed by CRISPR/Cas9 and ssODN in human cells. Journal of Visualized Experiments. (126), (2017).
- Nandal, A., Mallon, B., Telugu, B. P. Efficient generation and editing of feeder-free IPSCs from human pancreatic cells using the CRISPR-Cas9 system. Journal of Visualized Experiments. (129), (2017).
- Mohr, S. E., Hu, Y., Ewen-Campen, B., Housden, B. E., Viswanatha, R., Perrimon, N. CRISPR guide RNA design for research applications. The FEBS Journal. 283 (17), 3232-3238 (2016).
- Bauer, D. E., Canver, M. C., Orkin, S. H. Generation of genomic deletions in mammalian cell lines via CRISPR/Cas9. Journal of Visualized Experiments. (95), e52118 (2015).
- Hsu, P. D., Scott, D. A., et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nature Biotechnology. 31 (9), 827-832 (2013).
- Heigwer, F., Kerr, G., Boutros, M. E-CRISP: fast CRISPR target site identification. Nature Methods. 11 (2), 122-123 (2014).
- Moreno-Mateos, M. A., Vejnar, C. E., et al. CRISPRscan: designing highly efficient sgRNAs for CRISPR-Cas9 targeting in vivo. Nature Methods. 12 (10), 982-988 (2015).
- Labun, K., Montague, T. G., Gagnon, J. A., Thyme, S. B., Valen, E. CHOPCHOP v2: a web tool for the next generation of CRISPR genome engineering. Nucleic Acids Research. 44, 272-276 (2016).
- Haeussler, M., Schönig, K., et al. Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biology. 17 (1), 148 (2016).
- Lo, T. -W., Pickle, C. S., et al. Precise and heritable genome editing in evolutionarily diverse nematodes using TALENs and CRISPR/Cas9 to engineer insertions and deletions. Genetics. 195 (2), 331-348 (2013).
- Bassett, A., Liu, J. -L. CRISPR/Cas9 mediated genome engineering in Drosophila. Methods. 69 (2), 128-136 (2014).
- Prior, H., Jawad, A. K., MacConnachie, L., Beg, A. A. Highly efficient, rapid and co-CRISPR independent genome editing in Caenorhabditis elegans. G3: Genes, Genomes, Genetics. , Bethesda, MD. (2017).
- Hirsh, A. Cas9 expression and purification protocol. protocols.io. , (2017).
- DeWitt, M. A., Wong, J. In vitro transcription of guide RNAs. protocols.io. , (2017).
- Yang, L., Guell, M., et al. Optimization of scarless human stem cell genome editing. Nucleic Acids Research. 41 (19), 9049-9061 (2013).
- Richardson, C. D., Ray, G. J., DeWitt, M. A., Curie, G. L., Corn, J. E. Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA. Nature Biotechnology. 34 (3), 339-344 (2016).
- Paix, A., Folkmann, A., Seydoux, G. Precision genome editing using CRISPR-Cas9 and linear repair templates in C. elegans. Methods. 121-122, 86-93 (2017).
- Mello, C., Fire, A. DNA transformation. Methods in Cell Biology. 48, 451-482 (1995).
- Sutter Pipette Cookbook. , Available from: https://www.sutter.com/PDFs/pipette_cookbook.pdf (2017).
- Stiernagle, T. Maintenance of C. elegans. WormBook: the online review of C. elegans biology. , (2006).
- Evans, T. C. Transformation and microinjection. WormBook: the online review of C. elegans biology. , (2006).
- Berkowitz, L. A., Knight, A. L., Caldwell, G. A., Caldwell, K. A. Generation of stable transgenic C. elegans using microinjection. Journal of Visualized Experiments. (18), (2008).
- Kim, H., Ishidate, T., et al. A co-CRISPR strategy for efficient genome editing in Caenorhabditis elegans. Genetics. 197 (4), 1069-1080 (2014).
- Arribere, J. A., Bell, R. T., Fu, B. X. H., Artiles, K. L., Hartman, P. S., Fire, A. Z. Efficient marker-free recovery of custom genetic modifications with CRISPR/Cas9 in Caenorhabditis elegans. Genetics. 198 (3), 837-846 (2014).
- Ward, J. D. Rapid and precise engineering of the Caenorhabditis elegans genome with lethal mutation co-conversion and inactivation of NHEJ repair. Genetics. 199 (2), 363-377 (2015).
- Farboud, B., Meyer, B. J. Dramatic enhancement of genome editing by CRISPR/Cas9 through improved guide RNA design. Genetics. 199 (4), 959-971 (2015).
- Wood, A. J., Lo, T. -W., et al. Targeted genome editing across species using ZFNs and TALENs. Science. 333 (6040), 307 (2011).
- Friedland, A. E., Tzur, Y. B., Esvelt, K. M., Colaiácovo, M. P., Church, G. M., Calarco, J. A. Heritable genome editing in C. elegans via a CRISPR-Cas9 system. Nature Methods. 10 (8), 741-743 (2013).
- Dickinson, D. J., Ward, J. D., Reiner, D. J., Goldstein, B. Engineering the Caenorhabditis elegans genome using Cas9-triggered homologous recombination. Nature Methods. 10 (10), 1028-1034 (2013).
- Rehm, E. J., Hannibal, R. L., Chaw, R. C., Vargas-Vila, M. A., Patel, N. H. Injection of Parhyale hawaiensis blastomeres with fluorescently labeled tracers. Cold Spring Harbor Protocols. 2009 (1), (2009).
- Gerberding, M., Browne, W. E., Patel, N. H. Cell lineage analysis of the amphipod crustacean Parhyale hawaiensis reveals an early restriction of cell fates. Development (Cambridge, England). 129 (24), 5789-5801 (2002).
- Browne, W. E., Price, A. L., Gerberding, M., Patel, N. H. Stages of embryonic development in the amphipod crustacean, Parhyale hawaiensis. Genesis. 42 (3), New York, NY. 124-149 (2005).
- Rehm, E. J., Hannibal, R. L., Chaw, R. C., Vargas-Vila, M. A., Patel, N. H. Fixation and dissection of Parhyale hawaiensis embryos. Cold Spring Harbor Protocols. 2009 (1), (2009).
- Rehm, E. J., Hannibal, R. L., Chaw, R. C., Vargas-Vila, M. A., Patel, N. H. In situ hybridization of labeled RNA probes to fixed Parhyale hawaiensis embryos. Cold Spring Harbor Protocols. 2009 (1), (2009).
- Rehm, E. J., Hannibal, R. L., Chaw, R. C., Vargas-Vila, M. A., Patel, N. H. Antibody staining of Parhyale hawaiensis embryos. Cold Spring Harbor Protocols. 2009 (1), (2009).
- Kontarakis, Z., Pavlopoulos, A. Transgenesis in non-model organisms: the case of Parhyale. Methods in Molecular Biology. 1196, Clifton, NJ. 145-181 (2014).
- Kim, H. J., Lee, H. J., Kim, H., Cho, S. W., Kim, J. -S. Targeted genome editing in human cells with zinc finger nucleases constructed via modular assembly. Genome Research. 19 (7), 1279-1288 (2009).
- Qiu, P., Shandilya, H., D'Alessio, J. M., O'Connor, K., Durocher, J., Gerard, G. F. Mutation detection using Surveyor nuclease. BioTechniques. 36 (4), 702-707 (2004).
- Brinkman, E. K., Chen, T., Amendola, M., van Steensel, B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Research. 42 (22), 168 (2014).
- Tsai, S. Q., Zheng, Z., et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nature Biotechnology. 33 (2), 187-197 (2015).
- Frock, R. L., Hu, J., Meyers, R. M., Ho, Y. -J., Kii, E., Alt, F. W. Genome-wide detection of DNA double-stranded breaks induced by engineered nucleases. Nature Biotechnology. 33 (2), 179-186 (2015).
- Smith, C., Gore, A., et al. Whole-genome sequencing analysis reveals high specificity of CRISPR/Cas9 and TALEN-based genome editing in human iPSCs. Cell Stem Cell. 15 (1), 12-13 (2014).
- Veres, A., Gosis, B. S., et al. Low incidence of off-target mutations in individual CRISPR-Cas9 and TALEN targeted human stem cell clones detected by whole-genome sequencing. Cell Stem Cell. 15 (1), 27-30 (2014).
- Kim, D., Bae, S., et al. Digenome-seq: genome-wide profiling of CRISPR-Cas9 off-target effects in human cells. Nature Methods. 12 (3), 237-243 (2015).
- Hendel, A., Fine, E. J., Bao, G., Porteus, M. H. Quantifying on- and off-target genome editing. Trends in Biotechnology. 33 (2), 132-140 (2015).
- O'Geen, H., Yu, A. S., Segal, D. J. How specific is CRISPR/Cas9 really. Current Opinion in Chemical Biology. 29, 72-78 (2015).
- Tsai, S. Q., Joung, J. K. Defining and improving the genome-wide specificities of CRISPR-Cas9 nucleases. Nature Reviews Genetics. 17 (5), 300-312 (2016).
- Hoban, M. D., Cost, G. J., et al. Correction of the sickle cell disease mutation in human hematopoietic stem/progenitor cells. Blood. 125 (17), 2597-2604 (2015).
- Simeonov, D. R., Gowen, B. G., et al. Discovery of stimulation-responsive immune enhancers with CRISPR activation. Nature. , (2017).
- Hultquist, J. F., Schumann, K., et al. A Cas9 ribonucleoprotein platform for functional genetic studies of HIV-host interactions in primary human T cells. Cell Reports. 17 (5), 1438-1452 (2016).
- Paix, A., Wang, Y., et al. Scalable and versatile genome editing using linear DNAs with microhomology to Cas9 sites in Caenorhabditis elegans. Genetics. 198 (4), 1347-1356 (2014).
- Lee, K., Mackley, V. A., et al. Synthetically modified guide RNA and donor DNA are a versatile platform for CRISPR-Cas9 engineering. eLife. 6, (2017).
- Minkenberg, B., Wheatley, M., Yang, Y. CRISPR/Cas9-enabled multiplex genome editing and its application. Progress in Molecular Biology and Translational Science. 149, 111-132 (2017).
- Doench, J. G., Fusi, N., et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nature Biotechnology. 34 (2), 184-191 (2016).
- Doench, J. G., Hartenian, E., et al. Rational design of highly active sgRNAs for CRISPR-Cas9-mediated gene inactivation. Nature Biotechnology. , 1-8 (2014).
- Liu, H., Wei, Z., Dominguez, A., Li, Y., Wang, X., Qi, L. S. CRISPR-ERA: a comprehensive design tool for CRISPR-mediated gene editing, repression and activation. Bioinformatics (Oxford, England). 31 (22), 3676-3678 (2015).
- Wu, X., Scott, D. A., et al. Genome-wide binding of the CRISPR endonuclease Cas9 in mammalian cells. Nature Biotechnology. 32 (7), 670-676 (2014).
- Bogenhagen, D. F., Brown, D. D. Nucleotide sequences in Xenopus 5S DNA required for transcription termination. Cell. 24 (1), 261-270 (1981).
- Cozzarelli, N. R., Gerrard, S. P., Schlissel, M., Brown, D. D., Bogenhagen, D. F. Purified RNA polymerase III accurately and efficiently terminates transcription of 5S RNA genes. Cell. 34 (3), 829-835 (1983).
- Chen, B., Gilbert, L. A., et al. Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell. 155 (7), 1479-1491 (2013).
- Gagnon, J. A., Valen, E., et al. Efficient mutagenesis by Cas9 protein-mediated oligonucleotide insertion and large-scale assessment of single-guide RNAs. PLoS ONE. 9 (5), 98186 (2014).
- Ren, X., Yang, Z., et al. Enhanced specificity and efficiency of the CRISPR/Cas9 system with optimized sgRNA parameters in Drosophila. Cell Reports. 9 (3), 1151-1162 (2014).
- Ran, F. A., Hsu, P. D., Wright, J., Agarwala, V., Scott, D. A., Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nature Protocols. 8 (11), 2281-2308 (2013).
- Serano, J. M., Martin, A., et al. Comprehensive analysis of Hox gene expression in the amphipod crustacean Parhyale hawaiensis. Developmental Biology. 409 (1), 297-309 (2016).
- Sternberg, S. H., Redding, S., Jinek, M., Greene, E. C., Doudna, J. A. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature. , 1-17 (2014).
- Lee, K., Conboy, M., et al. Nanoparticle delivery of Cas9 ribonucleoprotein and donor DNA in vivo induces homology-directed DNA repair. Nature Biomedical Engineering. 1 (11), 889-901 (2017).
