

Summary
Enantiomerically angereicherten Bispiro [γ-Butyrolacton-Pyrrolidin-4, 4'-Pyrazolone] Skelette sind asymmetrisch durch eine einfache Organocatalytic 1,3-dipolare Cycloaddition Reaktion synthetisiert.
Abstract
Bispirocyclic Gerüste sind eines der wichtigen strukturellen Untereinheiten in vielen Naturprodukten, die vielfältige und attraktive biologische Aktivitäten aufweisen. Vor kurzem haben wir eine effiziente Organocatalytic Strategie entwickelt bietet einfache Zugang zu einer Vielzahl von enantiomerically angereicherten Bispiro [γ-Butyrolacton-Pyrrolidin-4, 4'-Pyrazolone] Skelette. In diesem Beitrag zeigen wir Ihnen ein detailliertes Protokoll für die asymmetrische Synthese von Drogen wie Bispirocyclic Verbindungen mit zwei Spirocyclic-Kohlenstoff-Zentren über eine Organocatyltic 1,3-dipolare Cycloaddition Reaktion. Spirocyclization Synthone α-imino γ-Lactone und Alkylidene Pyrazolones bereit sind erstens die unterliegen dann einer Cycloaddition Reaktion in Anwesenheit einer bifunktionelle Squaramide Organocatalyst, die gewünschte Bispirocycles in hohen Ausbeuten zu leisten und ausgezeichnete Stereoselectivities. Chirale Hochleistungs-Flüssigkeitschromatographie (HPLC) wird durchgeführt, um die Enantiomeren Reinheit der Produkte zu bestimmen, und der d.r.-Wert wird von Proton kernmagnetische Resonanz (1H NMR) geprüft. Die absolute Konfiguration des Produkts wird nach einer kristallographischen Röntgenstrahlanalyse zugewiesen. Diese synthetischen Strategie ermöglicht es Wissenschaftlern, eine Vielfalt an Bispirocyclic Gerüste in hohen Ausbeuten und exzellenten Diastereo und Enantioselektivitäten vorzubereiten.
Introduction
Chirale Spirocyclic Verbindungen gefunden in Naturprodukten, chiraler Liganden verbreitet und metallorganischen komplexe entstanden als attraktive synthetische Ziele aufgrund ihrer strukturellen Komplexität und biologische Aktivität1,2, 3. Insbesondere sind Bispirocyclic Gerüste, gekennzeichnet durch drei Ringe mit zwei starren Spirocenters, strukturelle Untereinheiten in vielen Naturprodukten mit wichtigen biologischen Aktivitäten4,5. Infolgedessen hat die Konstruktion von Verbindungen mit Stereocontrolled, optisch reinen Bispirocyclic Skelette in den letzten Jahrzehnten große hingewiesen. Eine große Anzahl von Spirocyclic Verbindungen und deren Derivate wurden erfolgreich durch metallorganische Ansätze synthetisiert und Organocatalytic nähert, z. B. asymmetrische Cycloadditions wie 1,3-dipolare Cycloadditions und Diels-Alder Reaktionen6,7,8. Diese Moleküle sind jedoch meist Monospirocyclic Strukturen, während Bispirocyclic Strukturen sind weniger berichtete über und beschränkt sich auf den Bau von Indol-basierten Bispirocycles.
Um mehr strukturell vielfältigen Bispirocyclic Verbindungen zu erhalten, wurde die Vielseitigkeit der Cycloaddition Synthone für die asymmetrische Bauweise der Spirocyclic Zentren erforscht9,10,11. Vor allem mit bifunktionelle Squaramide Organokatalysatoren, Azomethine Ylide12,13,14, z. B. α-imino γ-Lactone und Dipolarophiles, wie z. B. Alkylidene Pyrazolones15,16 ,17, sind in der Lage, eine einfache 1,3-dipolare Cycloaddition konstruieren Bispirocyclic Skelette mit mehreren Stereozentren, so dass sie die perfekte Spirocyclization Synthone (Abbildung 1) zu unterziehen. Nachdem die Optimierung der Struktur des Organocatalyst und Reaktionslösungsmittel, diese Cycloaddition Prozess effizient das gewünschte Produkt mit hohen Erträgen und exzellenten Enantio und Diastereoselectivity bietet. Darüber hinaus zeigt diese Reaktion eine relativ hohe strukturelle Toleranz auf ein breites Spektrum von deutscher Synthone mit unterschiedlichen Funktionsgruppen18. Diese neue Methode bietet einen effizienten Zugriff auf eine Vielzahl von hoch funktionalisierten Drogen wie Verbindungen mit zwei quartären Spirocenters über eine einfache Organocatalytic Cycloaddition, leuchtende Lichter auf seine Anwendung in der strukturellen Vielfalt orientierten die Synthese dieser interessanten Klasse von Verbindungen.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
Achtung: Bitte konsultieren Sie alle relevanten Sicherheitsdatenblätter (SDB) vor dem Gebrauch. Chemikalien und Lösungsmitteln verwendet Reagens Klasse waren und wurden ohne weitere Reinigung verwendet. Alle Reaktionen, die Luft oder feuchteempfindlicher Reagenzien oder Zwischenprodukte wurden unter Argon Atmosphäre durchgeführt.
1. Vorbereitung des α-Arylidiene Pyrazolinone Arten
-
Vorbereitung der pyrazolones
- Eine 250 mL Rundboden Flasche 40 mL Eisessig aus einem Messzylinder bei Raumtemperatur hinzufügen. Rühren Sie die Lösung unter Zugabe von Hydrazin (1 Äquivalent, 1,58 Mol/L) und Methyl-Acetate (1 Äquivalent, 1,58 Mol/L). Statten Sie den Kolben mit einem Reflux-Kondensator.
Hinweis: Diese Konzentration wird verwendet, weil eine geringere Konzentration zu einer langsameren Reaktion Rate führt. - Hitze der Reaktionskolben bis 120 ° C in einem Ölbad unter ständigem Rühren 3 h. Nach dem Abkühlen der Reaktionskolben auf Umgebungstemperatur, entfernen Sie den magnetischen Stir bar, mit einem Stir Bar Retriever. Konzentrieren sich die Reaktionsmischung mit einem Drehverdampfer bei 60 ° C. Verschütten des Reaktionsgemisches wegen Unterdruck zu vermeiden.
- Der Reaktionskolben 20 mL entionisiertem Wasser hinzu und übertragen Sie die Lösung in einem separatory Trichter. Extrahieren die wässrige Schicht 3 X mit Ethylacetat (30 mL). Kombinieren Sie die organischen Schichten im separatory Trichter und waschen Sie sie 2 X mit Sole (50 mL).
- Trocknen Sie die kombinierten organischen Schichten über wasserfreiem Natriumsulfat für 1 h und dann entfernen Sie das Natriumsulfat durch Schwerkraft Filtration.
- Entfernen des Lösungsmittels auf einen Drehverdampfer bei vermindertem Druck und bei 35 ° C.
- Nach dem Entfernen des Lösungsmittels, gelten Sie die Pyrazolone-Arten bei der Durchführung von Abschnitt 4.
- Eine 250 mL Rundboden Flasche 40 mL Eisessig aus einem Messzylinder bei Raumtemperatur hinzufügen. Rühren Sie die Lösung unter Zugabe von Hydrazin (1 Äquivalent, 1,58 Mol/L) und Methyl-Acetate (1 Äquivalent, 1,58 Mol/L). Statten Sie den Kolben mit einem Reflux-Kondensator.
-
Vorbereitung der Α-Arylidiene Pyrazolinone
- Pyrazolone (1 Äquivalent, 0,49 Mol/L), Benzaldehyd (1 Äquivalent, 0,49 Mol/L), Magnesiumoxid (0,5 g, entspricht 0,6) und eine magnetische Stir Bar in einem Ofen getrocknet 100 mL Rundboden Kolben unter N2 Atmosphäre hinzufügen.
- Reaktionskolben, mit einer luftdichten Spritze wasserfreie Acetonitril (40 mL) hinzu und dann statten Sie die Flasche mit einem Reflux-Kondensator aus. Hitze der Reaktionskolben bis 120 ° C in einem Ölbad unter ständigem Rühren 12 h.
- Überwachen Sie den Fortschritt der Reaktion durch Dünnschichtchromatographie (TLC), mit Petroleum Ether: Ethylacetat (2:1 [V/V], Aufbewahrung Faktor Rf = 0,86) als ein Eluent.
- Nach der vollständigen Einnahme der Pyrazolone cool Reaktionskolben bis auf Raumtemperatur. Filtern von Magnesiumoxid durch eine Celite-Stecker.
- Entfernen Sie die überschüssige Acetonitril mit Drehverdampfer unter vermindertem Druck und bei 35 ° C. Reinigen Sie die Rückstände durch Säulenchromatographie an Kieselgel eluierenden mit Petroleum Ether: Ethylacetat (10:1 bis 8:1 [V/V]) um das Rohprodukt zu bieten.
- Fügen Sie das Rohprodukt in einem 100 mL Erlenmeyerkolben, ausgestattet mit einer magnetischen Stir Bar hinzu, und fügen Sie ein Mindestvolumen von 95 % Ethanol. Legen Sie den Kolben auf einer heißen Platte und zum sanften Kochen bringen Sie, bis der gesamte Volumenkörper nur aufgelöst ist. Nehmen Sie die Flasche aus der heißen Platte und kühlen Sie es ohne jede Erregung langsam ab.
Hinweis: Wenn die Mischung auf Raumtemperatur abgekühlt ist, wird die entsprechende α-Arylidiene-Pyrazolinone als reine Kristalle gebildet.
2. Synthese von α-imino γ-Lactone Arten
- Hinzufügen von α-amino-γ-Butyrolacton Hydrobromide (1 Äquivalent, 0,41 Mol/L), Magnesiumsulfat (1 Äquivalent, 0,41 Mol/L), Triethylamin (1 Äquivalent, 0,41 Mol/L) und eine magnetische Stir Bar in einem Ofen getrocknet 100 mL Rundboden Kolben unter N2 Atmosphäre.
- Reaktionskolben, mit einer luftdichten Spritze fügen Sie 36 mL wasserfreiem Dichlormethan hinzu. Rühren Sie die Reaktionsmischung bei Raumtemperatur 1 h. Hinzufügen der entsprechenden Thiophen-2-Carbaldehyde (1,1 Äquivalent, 0,45 Mol/L) zur Lösung und rühren Sie für ein anderes 12 h.
- Überwachen Sie den Fortschritt der Reaktion von TLC, mit Petroleum Ether: Ethylacetat (4:1 [V/V]) als ein Laufmittel bis vollständigen Verbrauch der Gattung Lacton aufgetreten ist, und dann Filter aus der Reaktionsmischung, mit einem Filterpapier mit einer Porengröße von 30−50 μm.
- Die resultierende Mischung 5 mL entionisiertem Wasser hinzu und trennen Sie die organische Schicht aus der wässrigen Phase zu. Extrahieren die wässrige Phase 2 X mit Dichlormethan (30 mL). Kombinieren Sie die organischen Schichten im separatory Trichter und waschen Sie sie 2 X mit Sole (50 mL).
- Trocknen Sie die kombinierten organischen Schichten über wasserfreiem Natriumsulfat für 1 h und dann entfernen Sie das Natriumsulfat durch Schwerkraft Filtration. Entfernen des Lösungsmittels auf einen Drehverdampfer bei vermindertem Druck und bei 35 ° C.
- Nach dem Entfernen des Lösungsmittels, gelten Sie α-Imino γ-Lactone Arten bei der Durchführung von Abschnitt 4.
3. Synthese von bifunktionelle Squaramide Katalysator C519
Hinweis: Für die Synthese von Organokatalysatoren 5C, siehe Abbildung 2.
-
Vorbereitung des 3-((3,5-bis(trifluoromethyl)phenyl)amino)-4-methoxycyclobut-3-ene-1,2-dione (Verbindung 1)
- 3,4-dimethoxycyclobut-3-ene-1,2-dione (1 Äquivalent, 0,63 Mol/L), 3,5-Bis (Trifluormethyl) Anilin (1.1 Äquivalent, 0,69 Mol/L), 20 mL Methanol und eine magnetische Stir Bar in einem Ofen getrocknet 100 mL Rundboden Kolben unter N2 Atmosphäre hinzufügen.
- Rühren Sie die Mischung bei Raumtemperatur für 48 h. Die Bildung des gelben Niederschlags ist Indiz dafür, dass die Reaktion stattfindet.
- Filter der Reaktionslösung durch einen Trichter ausgestattet mit Filterpapier und waschen das feste Produkt 3 X mit Methanol (15 mL). Trocknen Sie die gelbliche Lösung im Vakuum über Nacht um die Endprodukte als gelbliche Lösung leisten.
-
Synthese des Katalysators C5
- Fügen Sie 3-((3,5-bis(trifluoromethyl)phenyl)amino)-4-methoxycyclobut-3-ene-1,2-dione (Verbindung 1; 1 Äquivalent, 0,2 Mol/L) und (S)-(6-methoxyquinolin-4-yl)((1S,2R,4S,5R)-5-vinylquinuclidin-2-yl)methanamine (zusammengesetzte 2; 1 Äquivalent, 0,2 Mol/L) und eine magnetische Stir Bar in ein 25 mL Rundboden Kolben unter N2 Atmosphäre.
- Fügen Sie wasserfrei Dichlormethan (5 mL), mit einer luftdichten Spritze. Rühren Sie die Mischung bei Raumtemperatur für 48 h.
- Überwachen Sie den Fortschritt der Reaktion von TLC, mit Dichloromethane:methanol (10:1 [V/V], Rf = 0,49) als ein Eluent. Nachdem die Reaktion abgeschlossen ist, konzentrieren sich die Reaktionsmischung mit einem Drehverdampfer bei 40 ° C.
- Reinigen Sie die Rückstände durch Säulenchromatographie an Kieselgel eluierenden mit Dichloromethane:methanol (20:1 [V/V]), das gewünschte Produkt zu bieten.
4. asymmetrische Synthese Bispirocyclic Substanzen
- Trocken Sie eine 50 mL Rundboden Reaktionskolben mit einer magnetischen Stir Bar. Den Kolben aus dem Ofen nehmen und durch Einblasen drauf mit Inertgas vor Gebrauch auf Raumtemperatur abkühlen lassen.
- Α-Arylidiene-Pyrazolinone (1 Mmol, 1 entspricht, 0,1 Mol/L) und α-imino γ-Lactone (1,2 Mmol, 1,2 entspricht 0,12 Mol/L) in 50 mL Rundboden Kolben unter N2 Atmosphäre hinzufügen.
- Reaktionskolben, mit einer luftdichten Spritze fügen Sie wasserfreie Ethylether (10 mL hinzu). Dann fügen Sie die entsprechenden Organocatalyst (0,1 Äquivalent, 0,01 Mol/L hinzu) der Projektmappe und rühren Sie die Reaktionsmischung bei 40 ° C.
- Überwachen Sie den Fortschritt der Reaktion von TLC, mit Petroleum Ether: Ethylacetat (4:1 [V/V], Rf = 0,51) als ein Eluent.
Hinweis: Die Flecken von der Ausgangs- und Produkte wurden mit einer handgeführten 254 nm UV Lampe visualisiert. - Nach Abschluss die Reaktion konzentrieren Sie das Reaktionsgemisch, mit einem Drehverdampfer bei 40 ° C.
- Reinigen Sie die Rückstände durch Säulenchromatographie an Kieselgel eluierenden mit Petroleum Ether: Ethylacetat (4:1 [V/V]), das Endprodukt zu liefern.
- Das Endprodukt von 1H und 13C-NMR-Spektren, mit einem 400 MHz-NMR-Spektrometer zu charakterisieren. Die Ee-Werte des Produkts, mit einer chiralen Spalte zu bestimmen.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Untersucht wurden verschiedene Wasserstoffbrücken Spender bifunktionelle Organokatalysatoren in Anwesenheit von Organokatalysatoren in Dichlormethan (DCM) bei 25 ° C (Tabelle 1). Der repräsentative synthetischen Prozess der Organokatalysatoren ist in Abbildung 1dargestellt. Die Vorführung von verschiedenen Organokatalysatoren (Tabelle 1, Einträge 1-6) führte zu C5 mit ausgezeichneten Stereoselektivität (94 % Ee > d.r. 20:1, 5-Eintrag) und die beste Ausbeute (85 % Ausbeute). Eine weitere Optimierung der Lösungsmittel (Tabelle 1, Einträge 7-11) vorgeschlagen, dass Et2O war bei diesem synthetischen Verfahren vorzuziehen.
Deutlich, um die Allgemeingültigkeit der Reaktion zu untersuchen, eine Vielzahl von Substituenten von zwei Spirocyclization Synthone mit verschiedenen funktionellen Gruppen getestet wurden erfolgreich mit der optimierten Modell Reaktionsbedingungen, was in der gewünschten Bispirocycles mit guten bis sehr guten Ausbeuten und Stereoselektivität. Pyrazolinone 1a umfaßt einen Ersatz der Phenyl-Gruppe auf α-Arylidiene mit einer breiten Palette von Aryl, Naphthyl, und Thienyl Gruppen, Substrate mit verschiedenen Substituenten wie Ethyl, Decyl, Tert- Butyl und Benzyl-Gruppe 3- Position und Substituenten mit unterschiedlichen elektronischen Eigenschaften auf dem Aryl-Ring an 1-Position. Außerdem wurde das Substrat zur Erkundung Anwendungsbereich imino Lacton 2a, zyklische imino Ester glyko-mit einer 5-Methylthiophenyl, Phenyl oder 2-Naphthyl-Gruppe, bietet Bispirocycles mit ähnlichen Erträgen und erhebliche Stereoselectivities ersetzt.
Die Struktur der Bispirocyclic Produkte bestätigte 1H und 13C-NMR-Spektroskopie. Um die optische Reinheit und Stereoselektivität der Stereoisomer Produkte kennzeichnen, die Ee-Werte wurden ermittelt, mittels chirale HPLC und der d.r. Zeitwerte wurden von 400 MHz 1H NMR. Tadelnswerter HPLC Charakterisierung Ergebnisse von zusammengesetzten 3e sind in Abbildung 3gegeben. Zur Erkundung der strukturellen Relativitätstheorie diente Röntgenkristallographie 3e, enthüllt die absolute Konfiguration von Produkt- 3e (5S,R,R, 13R7 6) zu analysieren. Die Einkristall Struktur der 3e ist in Abbildung 4dargestellt. Hongkongs 1590396 enthält die kristallographischen Daten von 3e, die erreicht werden kann kostenlos von der Cambridge kristallographischen Data Centre (www.ccdc.cam.ac.uk/data_request/cif).
Zum Beispiel waren die Charakterisierungsdaten für Bispirocyclic Produkt (3e) wie folgt: Rf = 0,51 (4:1 [V/V], Petroleum Äther/EtOAc); 1 H NMR (400 MHz, CDCl3) δ 7,55 (d, J = 7,2 Hz, 2 H), 7,35-7,31 (m, 2 H), 7,26-7.22 (m, 2 H), 7.19-7.15 (m, 2 H), 6,98-6,93 (m, 3 H) 6.90-6,88 (m, 1 H), 5.03 (d, J = 11,6 Hz, 1 H), 4.57 (s, 1 H), 4,46-4.40 (m, 1 H), 4.09 (td, J = 8,8, 2,0 Hz 1 H), 3,80 (d, J = 11,6 Hz, 1 H), 2.67-2,61 (m, 1 H), 2.35-2,27 (m, 4 H); 13 C-NMR (100 MHz, CDCl3) δ 178,8, 171.9, 163,9, 161,4, 158,5, 137.1, 136,0, 131,3, 131,2, 128,8, 127,0, 125,8, 125,4, 125.0, 119,7, 116,3, 116,1, 70,8, 67,4, 66,0, 64.1, 57,5, 34,6, 13,6; 19 F NMR (376 MHz, CDCl3) δ 112,8. Der Ee-Wert wurde durch HPLC-Analytik, Hexan/2-Propanol 80/20, Durchflussmenge bestimmt = 1,0 mL/min, 254 nm, tr = 8,63 min (Major); HRMS (ESI) betr. C26H23N3O3SF+ [M + H]+ 476.1439, gefunden 476.1446.
| Eintrag | Katalysator | Lösungsmittel | Ertrag[ein[ ] (%) | d.r.[c] | EE[d] (%) |
| 1 | C1 | DCM | 81 | > 20:1 | 94 |
| 2 | C2 | DCM | 82 | 12:1 | 90 |
| 3 | C3 | DCM | 30 | 9:1 | 0 |
| 4 | C4 | DCM | 78 | > 20:1 | 71 |
| 5 | C5 | DCM | 85 | > 20:1 | 94 |
| 6 | C6 | DCM | 70 | 12:1 | 93 |
| 7 | C5 | Toluol | 79 | > 20:1 | 95 |
| 8 | C5 | THF | 73 | 15:1 | 89 |
| 9 | C5 | KCHL3 | 71 | > 20:1 | 93 |
| 10 | C5 | DCE | 81 | 18:1 | 91 |
| 11 | C5 | Et2O | 88 (83[b]) | > 20:1 | 98 |
|
[a] die Erträge wurden von 1H NMR-Analyse Rohprodukt mit 4-Iodoanisole als interner Standard bestimmt. [b] isolierten Ausbeute. [c] Verhältnis richtet sich nach 1H NMR. [d] festgelegt durch chirale HPLC. |
|||||
Tabelle 1: Optimierung der Reaktion Bedingung. Die Tabelle wurde von Chen Et Al.18geändert.

Abbildung 1: Modell Reaktion zwischen 1a und 2a. Strukturen der bifunktionelle Organokatalysatoren (C1-C6) werden aufgelistet. Diese Abbildung zeigt Reaktionen mit 1a (0,10 Mmol), 2a (0.12 Mmol) und Katalysator (10 Mol%) im Lösungsmittel (1 mL) bei Raumtemperatur für 8-72 h durchgeführt. Für experimentelle Einzelheiten siehe das Protokoll. Diese Zahl wurde von Chen Et Al.18geändert. Bitte klicken Sie hier für eine größere Version dieser Figur.
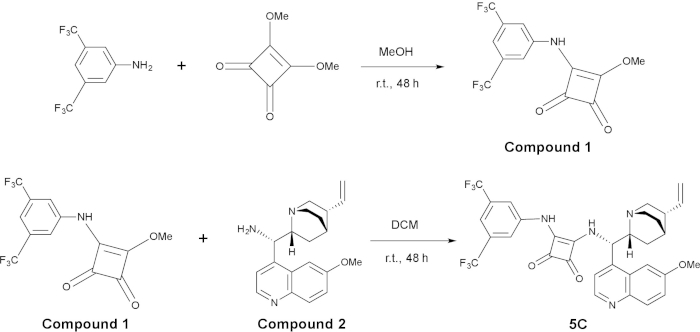
Abb. 2: Synthese von Organocatalyst 5C. Der obere Bereich ist die Synthese des zusammengesetzten 1, und die Bodenplatte ist die Synthese von 5 C aus zusammengesetzten 1 und zusammengesetzte 2. Bitte klicken Sie hier für eine größere Version dieser Figur.
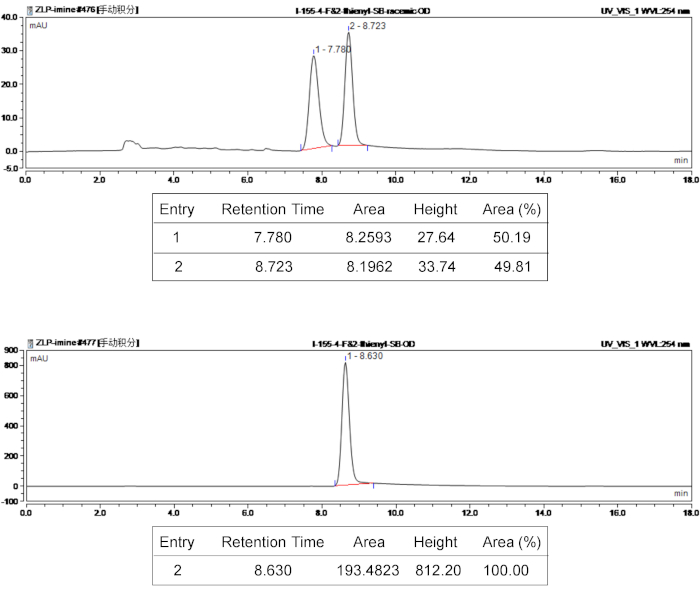
Abbildung 3: HPLC-Spektren von racemischem und chiralen Produkt 3e. Die Oberseite ist das HPLC-Spektrum von racemischem Produkt 3eund die Bodenplatte ist das HPLC-Spektrum von chiralen 3e. Bitte klicken Sie hier für eine größere Version dieser Figur.

Abbildung 4: Single-Kristallstruktur von 3e. Die linke Struktur ist die Einkristall Struktur von 3e, und die richtige Struktur ist 3e mit der Stereochemie von jedem Atom richtig benannt. Bitte klicken Sie hier für eine größere Version dieser Figur.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Die erfolgreiche Vorbereitung der Skelette, Bispiro [γ-Butyrolacton-Pyrrolidin-4, 4'-Pyrazolone] ist eine Reihe von Faktoren abhängig.
Der entscheidende Schritt von diesem Einschrittverfahren asymmetrische Cycloaddition ist die synergistical Aktivierung der α-Arylidiene Pyrazolinone 1a und zyklischen imino Ester 2a durch den bifunktionelle Squaramide Katalysator. Es wird durch die Bildung von mehreren intermolekulare Wasserstoffbrücken zwischen Katalysator als Wasserstoffbrücken Spender und zwei Reaktion Substrate erreicht. Dementsprechend stellte mit große sterische Behinderung, C5, aus all der Wasserstoffbrücken Spender bifunktionelle Organokatalysatoren gezeigt, die besten Stereoselektivität. Das bekannte Protokoll verwendet 10 Mol % Katalysator in der Modellreaktion. Außerdem gilt das Erfordernis der hohen Löslichkeit von Substraten und Katalysator. Infolgedessen hat die Verwendung von Et2O als optimale Lösungsmittel nicht nur sicher, dass Substrate und Katalysator vollständig aufgelöst sind bei Raumtemperatur aber, dass sie eine glatte Cycloaddition mit hohen Erträgen und Stereoselectivities sowie unterziehen. Vor allem würde Wasser im Reaktionssystem zu schlechte Stereoselektivität führen. Um eine gelungene Synthese zu gewährleisten, ist es wichtig, die Trockenheit der alle Reagenzien und Lösungsmittel prüfen vor Beginn der Reaktions.
Die Cycloaddition ist kompatibel mit einer Vielzahl von substituierten α-Arylidiene Pyrazolinone. Substituenten mit einer unterschiedlichen Aryl-Gruppe auf α-Arylidiene sind speziell, gut verträglich. Elektron ausscheidende Aryl Gruppen aufgrund ihrer erhöhten Electrophilicity bei 1,3-dipolare Cycloaddition, werden bevorzugt in Bezug auf die Erträge und Stereoselektivität. Außerdem ersetzt Substrate wie 3-Position, Ethyl, Decyl, Tert-Butyl, und Benzylbutylphthalat Gruppen und 1-Position, durch verschiedene elektronische Aryl Ringe funktionalisiert, sind sehr verträglich. Darüber hinaus sind die Substituenten des zyklischen imino Ester glyko-auf imino Lacton mit Phenyl, Thiophenyl oder Naphthyl Gruppen auch kompatibel mit der Reaktion. Es ist bemerkenswert, dass, um eine erfolgreiche Reaktion zu gewährleisten, einen kleinen Überschuss an imino Lacton (1.2 gleichwertig) erforderlich. In den meisten Fällen befinden sich die Konzentrationen der Substrate bei 0,1-0,12 Mol/L-Skala in 1 mL Lösungsmittel. Je nach Art der Substrate und Katalysatoren dauert die einstufige Cycloaddition Reaktion 8-72 h bei Raumtemperatur.
Es ist erwähnenswert, dass diese Cycloaddition, sofern ein hohes Maß an Stereoselektivität R4 Substituent imino Lacton 2 Thiolphenyl, 5-Methylthiophenyl oder 2-Naphthyl Gruppe war. Als mit anderen Alkyl Substituenten oder heterozyklische Substituenten R4 Substituenten ersetzt wurde, wurde jedoch eine niedrige Stereoselektivität oder eine geringe Reaktion Ausbeute erzielt.
Zusammenfassend lässt sich sagen ermöglicht die vorgestellte Protokoll die direkte asymmetrische Bauweise des Bispiro [γ-Butyrolacton-Pyrrolidin-4, 4'-Pyrazolone] mit einer effizienten einstufige Organocatalytic 1,3-dipolare Cycloaddition Reaktion in sehr guten Ausbeuten und ein hohes Maß an Stereoselektivität. Darüber hinaus diese neue Methode ist kompatibel mit zwei Synthone mit vielseitigen funktionellen Gruppen und für die Synthese von verschiedenen therapeutischen Wirkstoffen mit Bispirocyclic Gerüste nützlich sein sollte.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Die Autoren haben nichts preisgeben.
Acknowledgments
Die Autoren schätzen die finanzielle Unterstützung durch die National Natural Science Foundation of China (Nr. 21708051, X.C.).
Materials
| Name | Company | Catalog Number | Comments |
| Acetonitrile, anhydrous, 99.9% | Innochem (China) | A0080 | |
| α-amino-γ-butyrolactone hydrobromide, 98% | Alfa Aesar | B23148 | |
| 3,5-bis(trifluoromethyl)aniline, 98+% | Adamas | 48611B | |
| Dichloromethane, 99.5% | Greagent | G81014H | |
| 3,4-dimethoxycyclobut-3-ene-1,2-dione, 98+% | Leyan (China) | 1062550 | |
| Ethanol, 99.5% | Greagent | G73537B | |
| Ethyl acetate, 99.5% | Greagent | G23272L | |
| Ethyl ether,anhydrous,99.5% | Greagent | G69159B | |
| Ethyl 3-oxobutanoate, 98% | TCI | A0649 | |
| 4-fluorobenzaldehyde, 98% | Innochem (China) | A24295 | |
| Glacial acetic acid, 99.5% | Greagent | G73562B | |
| Magnesium oxide, 99+% | Alfa Aesar | 44733 | |
| Magnesium sulfate, 98% | Greagent | G80872C | |
| Methanol, 99.5% | Greagent | G75851A | |
| Petroleum ether | Greagent | G84208D | |
| Phenylhydrazine, 98% | Innochem (China) | A57671 | |
| (S)-(6-methoxyquinolin-4-yl)((1S,2R,4S,5R)-5-vinylquinuclidin-2-yl)methanamine | DAICEL Group | 111240 | |
| Sodium sulfate,anhydrous,99% | Greagent | G82667A | |
| Thiophene-2-carbaldehyde, 98% | J & K scientific (China) | 124605 | |
| Triethylamine, 99% | J & k scientific (China) | 432915 |
References
- Rios, R. Enantioselective methodologies for the synthesis of spiro compounds. Chemical Society Reviews. 41 (3), 1060-1074 (2012).
- Khan, R. K., et al. Synthesis, isolation, characterization, and reactivity of high-energy stereogenic-at-Ru carbenes: stereochemical inversion through olefin metathesis and other pathways. Journal of the American Chemical Society. 134 (30), 12438-12441 (2012).
- Wang, X., Han, Z., Wang, Z., Ding, K. Catalytic asymmetric synthesis of aromatic spiroketals by spinphox/iridium(I)-catalyzed hydrogenation and spiroketalization of alpha,alpha'-bis(2-hydroxyarylidene) ketones. Angewandte Chemie International Edition. 51 (4), 936-940 (2012).
- Kim, N., Sohn, M. J., Koshino, H., Kim, E. H., Kim, W. G. Verrulactone C with an unprecedented dispiro skeleton, a new inhibitor of Staphylococcus aureus enoyl-ACP reductase, from Penicillium verruculosum F375. Bioorganic & Medicinal Chemistry Letters. 24 (1), 83-86 (2014).
- Mulholland, D. A., Schwikkard, S. L., Crouch, N. R. The chemistry and biological activity of the Hyacinthaceae. Natural Product Reports. 30 (9), 1165-1210 (2013).
- Tan, B., Hernandez-Torres, G., Barbas, C. F. Highly efficient hydrogen-bonding catalysis of the Diels-Alder reaction of 3-vinylindoles and methyleneindolinones provides carbazolespirooxindole skeletons. Journal of the American Chemical Society. 133 (32), 12354-12357 (2011).
- Cayuelas, A., et al. Enantioselective Synthesis of Polysubstituted Spiro-nitroprolinates Mediated by a (R,R)-Me-DuPhos.AgF-Catalyzed 1,3-Dipolar Cycloaddition. Organic Letters. 18 (12), 2926-2929 (2016).
- Lacharity, J. J., et al. Total Synthesis of Unsymmetrically Oxidized Nuphar Thioalkaloids via Copper-Catalyzed Thiolane Assembly. Journal of the American Chemical Society. 139 (38), 13272-13275 (2017).
- Liu, K., Teng, H. L., Yao, L., Tao, H. Y., Wang, C. J. Silver-catalyzed enantioselective desymmetrization: facile access to spirolactone-pyrrolidines containing a spiro quaternary stereogenic center. Organic Letters. 15 (9), 2250-2253 (2013).
- Zhu, G., et al. Asymmetric [3 + 2] Cycloaddition of 3-Amino Oxindole-Based Azomethine Ylides and alpha,beta-Enones with Divergent Diastereocontrol on the Spiro[pyrrolidine-oxindoles]. Organic Letters. 19 (7), 1862-1865 (2017).
- Sun, W., et al. Organocatalytic diastereo- and enantioselective 1,3-dipolar cycloaddition of azlactones and methyleneindolinones. Angewandte Chemie International Edition. 52 (33), 8633-8637 (2013).
- Grigg, R., Kilner, C., Sarker, M. A. B., Orgaz de la Cierva, C., Dondas, H. A. X=Y–ZH compounds as potential 1,3-dipoles. Part 64: Synthesis of highly substituted conformationally restricted and spiro nitropyrrolidines via Ag(I) catalysed azomethine ylide cycloadditions. Tetrahedron. 64 (37), 8974-8991 (2008).
- Liu, T. L., He, Z. L., Tao, H. Y., Wang, C. J. Stereoselective construction of spiro(butyrolactonepyrrolidines) by highly efficient copper(I)/TF-BiphamPhos-catalyzed asymmetric 1,3-dipolar cycloaddition. Chemistry. 18 (26), 8042-8046 (2012).
- Wang, L., Shi, X. M., Dong, W. P., Zhu, L. P., Wang, R. Efficient construction of highly functionalized spiro[gamma-butyrolactone-pyrrolidin-3,3'-oxindole] tricyclic skeletons via an organocatalytic 1,3-dipolar cycloaddition. Chemical Communications. 49 (33), 3458-3460 (2013).
- Yetra, S. R., Mondal, S., Mukherjee, S., Gonnade, R. G., Biju, A. T. Enantioselective Synthesis of Spirocyclohexadienones by NHC-Catalyzed Formal [3+3] Annulation Reaction of Enals. Angewandte Chemie International Edition. 55 (1), 268-272 (2016).
- Liu, J. Y., Zhao, J., Zhang, J. L., Xu, P. F. Quaternary Carbon Center Forming Formal [3 + 3] Cycloaddition Reaction via Bifunctional Catalysis: Asymmetric Synthesis of Spirocyclohexene Pyrazolones. Organic Letters. 19 (7), 1846-1849 (2017).
- Mondal, S., Mukherjee, S., Yetra, S. R., Gonnade, R. G., Biju, A. T. Organocatalytic Enantioselective Vinylogous Michael-Aldol Cascade for the Synthesis of Spirocyclic Compounds. Organic Letters. 19 (16), 4367-4370 (2017).
- Chen, N., et al. Asymmetric Synthesis of Bispiro[γ-butyrolactone-pyrrolidin-4,4'-pyrazolone] Scaffolds Containing Two Quaternary Spirocenters via an Organocatalytic 1,3-Dipolar Cycloaddition. European Journal of Organic Chemistry. 2018 (23), 2939-2943 (2018).
- Yang, W., Du, D. M. Highly enantioselective Michael addition of nitroalkanes to chalcones using chiral squaramides as hydrogen bonding organocatalysts. Organic Letters. 12 (23), 5450-5453 (2010).
