In this project, a homogeneous glycoconjugate vaccine was prepared using the amber stop codon suppression strategy to introduce an UAA at a defined site (Figure 1). Pneumoccocal surface adhesin A was selected as the carrier protein moiety. This protein is highly conserved and expressed by all strains of Streptococcus pneumoniae22. It is highly immunogenic and previously used as a carrier in pneumococcal vaccine formulations21,23. As a proof-of-concept, the UAA propargyl-lysine efficiently charged by the wild type pyrrolysyl-tRNA synthetase (PylRS)/tRNA pair of the archaea Methanosarcina mazei was investigated. The propargyl-lysine is commercially available but can be advantageously prepared from Boc-L-lysine in only two synthetic steps (Figure 2). An amber codon was generated at a desired position in a pET24d plasmid containing the mPsaA gene. This plasmid was co-transformed with a pEVOL plasmid (a kind gift from Edward Lemke (EMBL19) containing orthogonal tools necessary to incorporate the propargyl-lysine, into competent E. coli BL21(DE3) strain. Positive co-transformed clones were selected using 25 µg/mL kanamycin and 30 µg/mL chloramphenicol. The plasmid pEVOL contains originally not one but two copies of the gene coding for MmPylRS to incorporate the propargyl-lysine residue: the first copy is under the control of a constitutive promoter while the expression of the other one is inducible in the presence of arabinose. However, we have noticed no dramatic decrease of propargyl-lysine incorporation if the MmPylRS gene under the control of constitutive promoter is suppressed.
The propargyl-lysine was introduced at position 32 in replacement of a lysine near the N-terminus of the PsaA. Any residue with a surface-exposed side-chain can virtually be exchanged in view of carrying out further conjugation. The mutated protein was produced in its mature form (mPsaAK32PrK) with inclusion of a cleavable 6-histidine tag sequence at its C-terminus. The efficiency of the mPsaAK32PrK production was checked by SDS-PAGE and Western Blot analysis using an anti-Histidine tag antibody, when growth was performed in the presence or the absence of the UAA propargyl-lysine and in comparison with the production of the wild type mPsaA (Figure 3). Visualization revealed a protein band at an expected molecular weight (Lanes 4, Figure 3A & 3B). The presence of a full-length protein strongly indicates the successful incorporation of the PrK into mPsaA. The intensity is, however, lower than that observed for wild type mPsaA (Lanes 2, Figure 3A & 3B). Leakage (i.e., production of the full-length protein without incorporation of the UAA) and premature release of the protein by the Release Factor RF1 during translation are two main drawbacks frequently encountered during this process. On one hand, no band at the expected molecular weight is visualized in the absence of propargyl-lysine meaning that no leakage occurred (Lanes 3, Figure 3A & 3B) and indirectly confirmed that the band observed on Lanes 4 corresponds to mPsaAK32PrK. On the other hand, no band can be seen at low molecular weight that could correspond to the truncated form of mPsaA (Lane 4 on Figure 3A). The mPsaAK32PrK was then purified by affinity chromatography, with a typical yield of 8 mg/L (in comparison to 12-20 mg/L for the wild type protein) and the incorporation of the propargyl-lysine residue was finally confirmed by mass spectrometry (Figure 4). The histidine tag was removed upon proteolytic cleavage using TEV protease (Figure 4). The stability of the mPsaAK32PrK thus obtained was assessed by circular dichroism, which showed that the structure of the protein was not disturbed by the mutation of the Lysine 32 into a propargyl-lysine (data not shown).
Having the mPsaAK32PrK, the reactivity of the alkyne for click chemistry was assessed using an azido-functionalized fluorescein and further used to conjugate a synthetic oligosaccharide antigen β-2-azidoethyl d-Galp-(1→4)-β-d-Glcp-(1→6)-[β-d-Galp-(1→4)]-β-d-GlcpNAc (Pn14TS) (Figure 5). This tetrasaccharide is related to the S. pneumoniae type 14 capsular polysaccharide and has previously been conjugated to mPsaA using different conjugation chemistries8,21,24. Experiments here were done in comparison with wild type mPsaA as a control. The histidine tag was first removed upon proteolytic cleavage using the TEV protease. The digested mPsaAK32PrK and mPsaA WT were then conjugated to the fluoroprobe (Figure 6A) or Pn14TS (Figure 6B). The reaction was assessed by SDS-PAGE. The small increase in the molecular weight of the sample between lane 6 and 7 (Figure 6B) indicates a successful conjugation with the tetrasaccharide Pn14TS. Finally, the glycoconjugate was purified by gel filtration and its identity confirmed by mass spectrometry (Figure 6C). The conjugation by click chemistry being quantitative the majority of the mPsaAK32PrK was conjugated with the Pn14TS-N3 as illustrated by the mass spectrometry results (Figure 6C).

Figure 1: Incorporation of propargyl-lysine (PrK) into mPsaA during translation using an orthogonal pyrrolysyl-tRNA synthetase/tRNA pair and TAG codon reassignment25. During translation, endogenous synthetases catalyze the link between amino acids and corresponding tRNAs. Then, loaded tRNAs are used by the ribosomal machinery to generate the neo-synthesized polypeptide. According to the amber stop codon suppression strategy, an orthogonal aminoacyl-tRNA synthetase (aaRS) (herein a pyrrolysyl-tRNA synthetase from M. mazei), loads an UAA (herein PrK) on its cognate tRNA which designed anticodon can read the amber stop codon (TAG) on the mRNA. This specific recognition directs the incorporation of the UAA into the specific site on the target protein. Figure reproduced from Wang et al.25. Please click here to view a larger version of this figure.

Figure 2: Propargyl-lysine synthesis. (A) Steps of propargyl-lysine synthesis. Insert: Monitoring of the deprotection of Boc-l-Lys(prop-2-ynyloxycarbonyl)-OH intermediate: thin-layer chromatography on 0.25 mm silica gel plates with fluorescent indicator (GF254) and visualised by charring with vanillin in sulfuric acid/ethanol (1.5:95 v/v); eluent: CH2Cl2/MeOH (9:1), left lane: Boc-l-Lys(prop-2-ynyloxycarbonyl)-OH (Rf 0.90), right lane: crude propargyl-lysine, (Rf 0.38). 400 MHz 1H (B) and 13C NMR spectra (C) of propargyl-lysine recorded in D2O. Please click here to view a larger version of this figure.
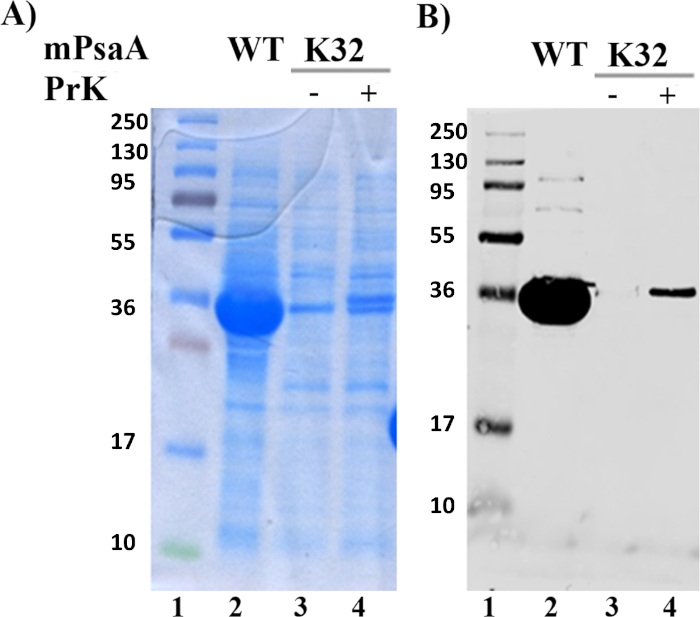
Figure 3: Analysis of crude cell samples. (A) SDS-PAGE analysis and (B) Western blot analysis on crude cell samples. Lane 1: unstained protein marker; Lane 2: crude cell extract of wild type mPsaA; Lane 3: crude cell extract of mPsaAK32TAG grown in the absence of PrK; Lane 4: crude cell extraction of mPsaAK32TAG grown in the presence of PrK. Conditions: 12% acrylamide gel, running at 100 V, 2 h. SDS-PAGE stained by Coomassie blue; Western Blot revealed using anti-histidine tag antibody and secondary antibody coupled with AlexaFluor680. Please click here to view a larger version of this figure.

Figure 4: Histidine tag removal and mass spectrometry analysis. (A) SDS-PAGE analysis. Lane 1: unstained protein marker; Lane 2: crude cell extract; Lane 3: unbound fraction; Lane 4: wash fraction with 10 mM imidazole; Lane 5: wash fraction with 20 mM imidazole; Conditions: 12% acrylamide gel running at 100 V for 2 h, and stained by Coomassie blue; (B) MALDI-TOF-MS spectra of (top) mPsaA WT, theoretical MW 33 103 Da, found 33 106 Da and (bottom) mPsaA K32PrK, theoretical 33 184 Da, found 33 192 Da. The found masses are within the expected margin error. Please click here to view a larger version of this figure.

Figure 5: Schematic representation of the conjugation strategy by click chemistry. A single tetrasaccharide bearing an azide is specifically coupled to its complementary biorthogonal alkyne group on mPsaA K32PrK (mPsaA representation based on the 1PSZ PDB file, with a resolution of 2.0 Å26). Please click here to view a larger version of this figure.

Figure 6: Histidine-tag digestion and conjugation of mPsaA with (A) fluorescein-N3 and with (B) Pn14TS. (A) SDS-PAGE Lanes 1-3: WT mPsaA; Lane 4-6: mPsaAK32PrK; (B) Lane 1: unstained protein marker; Lane 2-4: WT mPsaA; Lane 5-7: mPsaAK32PrK. 2 µg protein sample/lane, 12% acrylamide, 100 V, 2 h; (C) MALDI-TOF-MS spectra of the Pn14TS-mPsaAK32PrK theoretical MW 34 091 Da, found 34 088 Da. Please click here to view a larger version of this figure.
Supplemental File 1. Please click here to view this file (Right click to download).
