Source: Ewa Bukowska-Faniband1, Tilde Andersson1, Rolf Lood1
1 Département des sciences cliniques Lund, Division of Infection Medicine, Biomedical Center, Lund University, 221 00 Lund, Suède
La planète Terre est un habitat pour des millions d’espèces bactériennes, dont chacune a des caractéristiques spécifiques. L’identification des espèces bactériennes est largement utilisée dans l’écologie microbienne pour déterminer la biodiversité des échantillons environnementaux et la microbiologie médicale pour diagnostiquer les patients infectés. Les bactéries peuvent être classées à l’aide de méthodes de microbiologie conventionnelles, telles que la microscopie, la croissance sur des médias spécifiques, les tests biochimiques et sérologiques, et les tests de sensibilité aux antibiotiques. Au cours des dernières décennies, les méthodes de microbiologie moléculaire ont révolutionné l’identification bactérienne. Une méthode populaire est le séquençage du gène 16S ribosomal RNA (rRNA). Cette méthode est non seulement plus rapide et plus précise que les méthodes conventionnelles, mais permet également d’identifier les souches qui sont difficiles à cultiver en laboratoire. En outre, la différenciation des souches au niveau moléculaire permet la discrimination entre les bactéries phénotypiquement identiques (1-4).
16S rRNA se joint à un complexe de 19 protéines pour former une sous-unité 30S du ribosome bactérien (5). Il est encodé par le gène 16S rRNA, qui est présent et fortement conservé dans toutes les bactéries en raison de sa fonction essentielle dans l’assemblage de ribosome; cependant, il contient également des régions variables qui peuvent servir d’empreintes digitales pour des espèces particulières. Ces caractéristiques ont fait du gène 16S rRNA un fragment génétique idéal à utiliser dans l’identification, la comparaison et la classification phylogénétique des bactéries (6).
Le séquençage du gène rRNA 16S est basé sur la réaction en chaîne de polymérase (PCR) (7-8) suivie du séquençage de l’ADN (9). PCR est une méthode de biologie moléculaire utilisée pour amplifier des fragments spécifiques d’ADN à travers une série de cycles qui comprennent:
i) Dénaturation d’un modèle d’ADN à double brin
ii) Annealing des amorces (oligonucléotides courts) qui sont complémentaires au modèle
iii) Extension des amorces par l’enzyme de polymérase d’ADN, qui synthétise un nouveau brin d’ADN
Un aperçu schématique de la méthode est montré dans la figure 1.
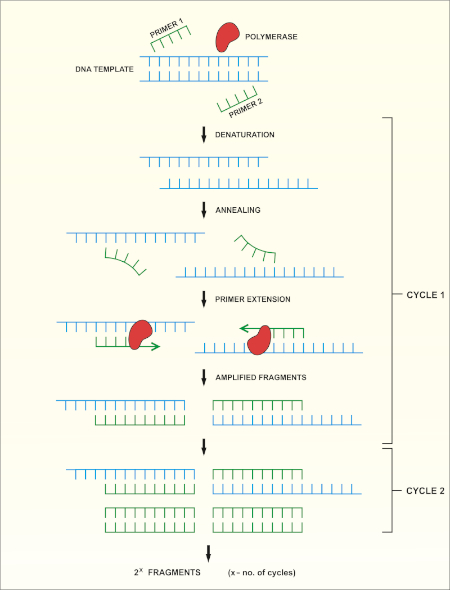
Figure 1 : Aperçu schématique de la réaction de PCR. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
Il y a plusieurs facteurs qui sont importants pour une réaction réussie de PCR, dont l’un est la qualité du modèle d’ADN. L’isolement de l’ADN chromosomique des bactéries peut être effectué à l’aide de protocoles standard ou de kits commerciaux. Une attention particulière doit être prise pour obtenir de l’ADN exempt de contaminants qui peuvent inhiber la réaction de PCR.
Les régions conservées du gène 16S rRNA permettent la conception de paires d’apprêt universels (un avant et un revers) qui peuvent se lier à la région cible et l’amplifier dans n’importe quelle espèce bactérienne. La région cible peut varier en taille. Alors que certaines paires d’amorces peuvent amplifier la plupart du gène 16S rRNA, d’autres n’amplifient que des parties de celui-ci. Des exemples d’amorces couramment utilisées sont présentés dans le tableau 1 et leurs sites de liaison sont représentés à la figure 2.
| Nom d’amorce | Séquence (5′-3′) | Avant/revers | référence |
| 8F b) | AGAGTTTGATCCTGGCTCAG | en avant | -1 |
| 27F (en) | AGAGTTTGATCMTGGCTCAG | en avant | -10 |
| 515F (en) | GTGCCAGCMGCCGCGGTAA | en avant | -11 |
| 911R | GCCCCCGTCAATTCMTTTGA | revenir sur | -12 |
| en 1391R | GACGCGGTGTGTRCA | revenir sur | -11 |
| 1492R | GGTTACCTTGTTACGACTT GGTTACCTTGTTACGACTT GGTTACCTTGTTACGACTT GGTTA | revenir sur | -11 |
Tableau 1 : Exemples d’oligonucléotides standard utilisés dans l’amplification des gènes 16S rRNA a).
a) Les longueurs prévues du produit PCR générées à l’aide des différentes combinaisons d’amorces peuvent être estimées en calculant la distance entre les sites de liaison pour l’avant et l’amorce inverse (voir la figure 2), par exemple la taille du PCR produit utilisant la paire d’apprêt 8F-1492R est de 1500 bp, et pour la paire d’apprêt 27F-911R 900 bp.
b) également connu sous le nom de fD1

Figure 2 : Figure représentative de la séquence de rRNA 16S et des sites de liaison d’amorce. Les régions conservées sont colorées en gris et les régions variables sont remplies de lignes diagonales. Pour permettre la plus haute résolution, l’amorce 8F et 1492R (nom basé sur l’emplacement sur la séquence d’ARNr) sont utilisées pour amplifier la séquence entière, permettant le séquençage de plusieurs régions variables du gène. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
Les conditions de cyclisme pour le PCR (c’est-à-dire la température et le temps requis pour que l’ADN soit dénaturé, annealed avec des amorces et synthétisé) dépendent du type de polymérase qui est utilisé et des propriétés des amorces. Il est recommandé de suivre les directives du fabricant pour une polymérase particulière.
Une fois le programme PCR terminé, les produits sont analysés par électrophoresis de gel d’agarose. Un PCR réussi donne une seule bande de taille prévue. Le produit doit être purifié avant le séquençage pour enlever les amorces résiduelles, les désoxyribonucleotides, la polymérase et le tampon qui étaient présents dans la réaction PCR. Les fragments d’ADN purifiés sont habituellement envoyés pour le séquençage aux services commerciaux de séquençage ; cependant, certaines institutions effectuent le séquençage de l’ADN dans leurs propres installations de base.
La séquence d’ADN est automatiquement générée à partir d’un chromatogramme d’ADN par un ordinateur et doit être soigneusement vérifiée pour la qualité, car l’édition manuelle est parfois nécessaire. Après cette étape, la séquence génétique est comparée aux séquences déposées dans la base de données rRNA 16S. Les régions de similitude sont identifiées, et les séquences les plus similaires sont livrées.