Source: Jonathan F. Blaize1, Elizabeth Suter1, et Christopher P. Corbo1
1 Département des sciences biologiques, Wagner College, 1 Campus Road, Staten Island NY, 10301
L’évaluation quantitative des procaryotes peut être onéreuse compte tenu de leur abondance, de leur propension à une prolifération exponentielle, de la diversité des espèces au sein d’une population et de leurs besoins physiologiques spécifiques. À ce défi s’ajoute la nature en quatre phases dans laquelle les bactéries se reproduisent (lag, journal, stationnaire et mort). La capacité d’estimer avec précision la concentration de micro-organismes est nécessaire pour réussir l’identification, l’isolement, la culture et la caractérisation (6). À ce titre, les microbiologistes ont utilisé la dilution en série et diverses techniques de placage depuis plus d’un siècle pour quantifier de façon fiable la charge bactérienne et virale dans les environnements cliniques, industriels, pharmaceutiques et universitaires de laboratoire (2,4,6). Les descriptions de cette méthodologie sont apparues pour la première fois en 1883 lorsque le scientifique et médecin allemand Robert Koch a publié ses travaux sur les agents pathogènes (2). Souvent appelées le père de la bactériologie moderne, les techniques prénommées de Koch sont devenues l’étalon-or de l’énumération des micro-organismes, culturables ou autres, dans le monde entier.
La dilution en série est une réduction systématique d’une entité connue ou inconnue (un soluté, un organisme, etc.) par une re-suspension successive d’une solution initiale (solution0) en volumes fixes d’un diluant liquide (blancs). Ces blancs se composent généralement de 0,45% salin, bien que la composition peut être variée (7). Bien qu’un expérimentateur puisse choisir n’importe quel volume pour chaque diluant, il s’agit le plus souvent d’un multiple de 10, ce qui facilite la réduction logarithmique de l’échantillon. Par exemple, la solution0 contient un total de 100 cellules E. coli suspendues dans 10 ml de bouillon nutritif. Si 1 ml de solution0 est enlevé et ajouté à 9 ml de solution saline (diluant1),la nouvelle solution (solution1) contiendrait 1/10e de la concentration initiale d’E. coli. Dans cet exemple, la nouvelle solution (solution1) contiendrait 10 cellules E. coli. Répéter ce processus en supprimant 1 ml de la solution1 et en l’ajoutant à 9 ml de salin (diluant2) donnerait la solution2, contenant seulement une seule cellule E. coli. Étant donné que chaque nouvelle solution (9 mL de diluant et 1 ml de solution) contient un total de 10 mL, nous pouvons conclure que le facteur de dilution pour cette réduction est de 10 ou qu’il s’agissait d’une dilution en série 10 fois(figure 1). Puisque nous avons seulement commencé avec 100 cellules dans cet exemple et nous diluons par un facteur de 10, seulement deux étapes sont nécessaires pour atteindre la concentration minimale absolue de 1 cellule.
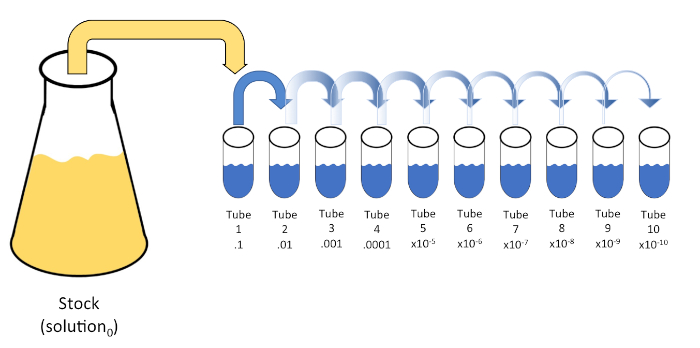
Figure 1 : Dilution en série d’une solution de stock. Un aliquot de 1 ml de la solution de stock (solution0) est ajouté au tube 1 qui contient 9 ml de 0,45% salin (dilent1); le produit de ce mélange est la solution1. Répétez en ajoutant 1 mL de la solution1 nouvellement créée et en l’ajoutant au tube 2. Aliquoting et la résuspension se poursuit de cette façon jusqu’à ce que le tube final est atteint, diluant la concentration du stock par un facteur de 10 chacun à chaque étape. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
La dilution en série est la technique la plus simple pour obtenir des concentrations gérables d’un organisme désiré et elle est complétée par des stries et des diffusions de boîtes de Pétri, seulement deux des nombreuses techniques de placage utilisées par les microbiologistes. Cet avantage de cette approche est que l’expérimentateur peut récolter des souches pures d’une seule espèce ou des souches séparées d’une population mixte (7). Le strage est accompli en introduisant un organisme à un milieu solide (généralement composé d’agarose) qu’il se développera sur si les éléments nutritifs appropriés sont disponibles. Balayer doucement une boucle d’inoculation stérile à travers le milieu (de sorte qu’une strie subtile reste) dans un modèle sinusoïdal rigide distribuera l’organisme proportionnellement à la fréquence de la forme d’onde de l’expérimentateur. Diviser le plat Petri en tiers ou en quarts (quadrant strie) et diminuer la fréquence de chaque strie à mesure qu’une nouvelle région du plat est entrée réduira graduellement le nombre de micro-organismes qui peuvent occuper cette région, produisant des colonies uniques au lieu d’un pelouse bactérienne non quantifiable. Le placage de propagation ne dilue pas en outre les échantillons; un épandeur de verre stérile est utilisé pour distribuer un aliquot de support de suspension sur un plat de pétri entier (figure 2). Les colonies qui poussent sur la plaque de propagation proviennent d’une seule cellule et chaque colonie sur le plat peut être comptée pour estimer le nombre d’unités de formation de colonies par millilitre (CFU) dans une suspension donnée, représentée comme CFU/mL (6) (Figure 3) Agar molle et réplique le placage sont des variations des techniques susmentionnées et permettent l’isolement du bactériophage et du criblage mutant, respectivement (1,7).
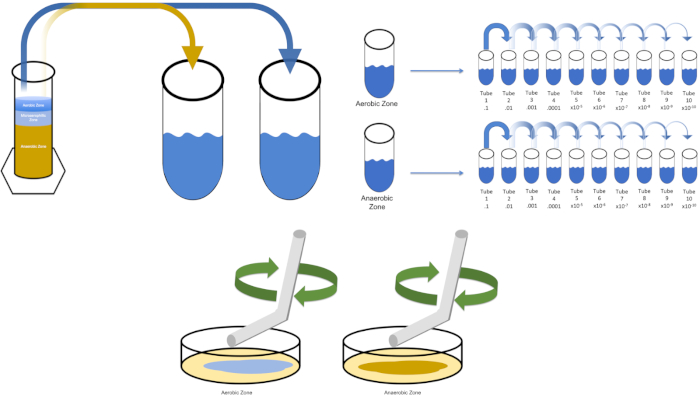
Figure 2 : Stries de plaques pour l’énumération bactérienne et l’isolement des souches. Étiquetez le fond d’un plat de pétri avec des informations d’identification (nom de la bactérie, date, média) et divisez-les en tiers. Après avoir sélectionné une dilution appropriée de l’échantillon de stock, prendre une boucle d’inoculation stérile (jetable ou enflammée) et la submerger le tube à essai (ici, T3). Soulevez légèrement le couvercle de la boîte de Pétri d’un côté de sorte que seule la boucle inoculation puisse accéder à l’agar. Glissez la boucle inoculée sur le dessus des médias d’une manière zig-zag en prenant soin de ne pas compromettre l’agar. Faites pivoter la plaque d’environ 1/3rd (118 degrés) et réduisez la fréquence des mouvements en zigzag. Tournez une dernière fois et réduisez une fois de plus la fréquence des zigzags. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
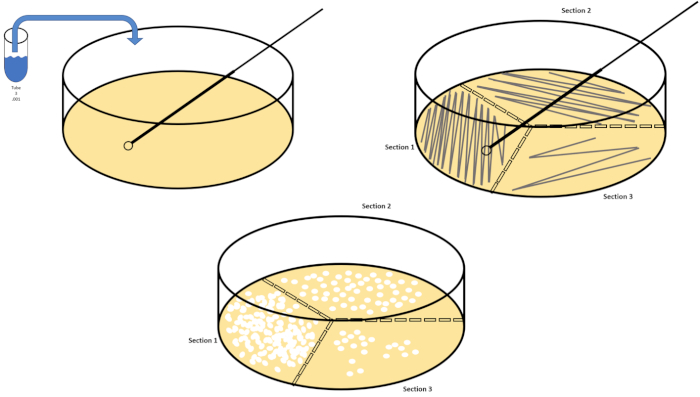
Figure 3 : Placage de propagation. 1 g de la zone aérobie a été resuspendu en T1, puis dilué en série. Un verre stérile ou une tige d’épandage jetable en plastique est utilisé pour distribuer de l’inoculum dans chaque plat. Ceci a été répété avec 1 g de la zone anaérobie. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
Comme pour les dilutions en série, une échelle logarithmique est utilisée pour exprimer la concentration de l’organisation. Le nombre de colonies cultivées dans des boîtes de Pétri standard mesurant 100 mm x15 mm peut être énuméré manuellement (ou automatisé à l’aide du traitement computationnel) en identifiant des grappes isolées de croissance. Les comptes qui totalisent moins de 30 ou plus de 300 devraient être définis comme trop peu nombreux à compter (TFTC) ou trop nombreux pour être comptés (TNTC), respectivement. Dans le cas de ce dernier, une dilution en série doit être effectuée pour réduire la concentration avant de restreaking un nouveau plat de pétri. La moyenne du nombre de colonies autonomes identifiées à partir de trois boîtes de Pétri distinctes et la multiplication de la moyenne par le facteur de dilution donneront cFU/mL; tracer le journal10 de CFU/mL contre le temps révélera le temps de génération moyen de l’organisme (7).