Source: Natalia Martin1, Andrew J. Van Alst1, Rhiannon M. LeVeque1, et Victor J. DiRita1
1 Département de microbiologie et de génétique moléculaire, Michigan State University
Les bactéries ont la capacité d’échanger du matériel génétique (acide déoxyribonucléique, ADN) dans un processus connu sous le nom de transfert horizontal de gènes. L’incorporation d’ADN exogène fournit un mécanisme par lequel les bactéries peuvent acquérir de nouveaux traits génétiques qui leur permettent de s’adapter aux conditions environnementales changeantes, telles que la présence d’antibiotiques ou d’anticorps (1) ou de molécules présentes dans les habitats naturels (2). Il existe trois mécanismes de transfert horizontal de gènes : la transformation, la transduction et la conjugaison (3). Ici, nous allons nous concentrer sur la transformation, la capacité des bactéries à prendre l’ADN libre de l’environnement. En laboratoire, le processus de transformation comporte quatre étapes générales : 1) Préparation de cellules compétentes, 2) Incubation de cellules compétentes avec ADN, 3) Récupération des cellules, et 4) Placage des cellules pour la croissance des transformateurs (figure 1).
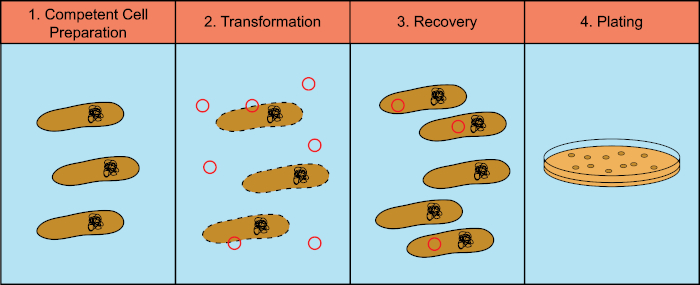
Figure 1 : Étapes générales du processus de transformation. Le processus de transformation comporte quatre étapes générales : 1) Préparation de cellules compétentes, 2) Incubation avec ADN, 3) Récupération des cellules et 4) Cellules de placage pour la croissance des transformateurs.
Pour que la transformation se produise, les bactéries récepteuses doivent être dans un état connu sous le nom de compétence. Certaines bactéries ont la capacité de devenir naturellement compétentes en réponse à certaines conditions environnementales. Cependant, beaucoup d’autres bactéries ne deviennent pas compétentes naturellement, ou les conditions pour ce processus sont encore inconnues. La capacité d’introduire l’ADN dans les bactéries a une gamme d’applications de recherche: pour générer plusieurs copies d’une molécule d’ADN d’intérêt, d’exprimer une grande quantité de protéines, comme un composant dans les procédures de clonage, et d’autres. En raison de la valeur de la transformation à la biologie moléculaire, il existe plusieurs protocoles visant à rendre les cellules artificiellement compétentes lorsque les conditions de compétence naturelle sont inconnues. Deux méthodes principales sont utilisées pour préparer des cellules artificiellement compétentes : 1) par le traitement chimique des cellules et 2) l’exposition des cellules aux impulsions électriques (électroporation). Le premier utilise différents produits chimiques selon la procédure pour créer une attraction entre l’ADN et la surface cellulaire, tandis que le second utilise des champs électriques pour générer des pores dans la membrane cellulaire bactérienne par laquelle les molécules d’ADN peuvent entrer. L’approche la plus efficace pour la compétence chimique est l’incubation avec des cations divalentes, plus particulièrement le calcium (Ca2)(4,5) La compétence induite par le calcium est la procédure qui sera décrite ici (6). Cette méthode est principalement utilisée pour la transformation des bactéries Gram-négatives, et qui sera au centre de ce protocole.
La procédure de transformation chimique implique une série d’étapes dans lesquelles les cellules sont exposées à des cations pour induire la compétence chimique. Ces étapes sont ensuite suivies d’un changement de température – choc thermique – qui favorise l’apport d’ADN étranger par la cellule compétente (7). Les enveloppes de cellules bactériennes sont chargées négativement. Chez les bactéries Gram-négatives comme Escherichia coli, la membrane externe est chargée négativement en raison de la présence de lipopolysaccharide (LPS) (8). Il en résulte la répulsion des molécules d’ADN de même charge négative. Dans l’induction chimique de compétence, les ions de calcium positivement chargés neutralisent cette répulsion de charge permettant l’absorption d’ADN sur la surface de cellules (9). Le traitement du calcium et l’incubation avec de l’ADN se font sur la glace. Par la suite, une incubation à des températures plus élevées (42 oC), le choc thermique, est effectuée. Ce déséquilibre de température favorise encore plus l’apport d’ADN. Les cellules bactériennes doivent être à la phase de croissance mi-exponentielle pour résister au traitement de choc thermique ; dans d’autres stades de croissance, les cellules bactériennes sont trop sensibles à la chaleur, ce qui entraîne une perte de viabilité qui diminue considérablement l’efficacité de la transformation.
Différentes sources d’ADN peuvent être utilisées pour la transformation. Typiquement, les plasmides, petites molécules circulaires d’ADN à double brin, sont utilisés pour la transformation dans la plupart des procédures de laboratoire dans E. coli. Pour que les plasmides soient maintenus dans la cellule bactérienne après la transformation, ils doivent contenir une origine de réplication. Cela leur permet d’être reproduits dans la cellule bactérienne indépendamment du chromosome bactérien. Toutes les cellules bactériennes ne se transforment pas au cours de la procédure de transformation. Ainsi, la transformation donne un mélange de cellules transformées et de cellules non transformées. Pour distinguer entre ces deux populations, une méthode de sélection pour identifier les cellules qui ont acquis le plasmide est utilisée. Les plasmides contiennent habituellement des marqueurs sélectionnables, qui sont des gènes codant un trait qui confère un avantage pour la croissance (c.-à-d. résistance à un antibiotique ou chimique ou sauvetage d’une auotrophie de croissance). Après la transformation, les cellules bactériennes sont plaquées sur des supports sélectifs, ce qui ne permet que la croissance des cellules transformées. Dans le cas des cellules transformées avec un plasmide conférant une résistance à un antibiotique donné, les médias sélectifs seront des supports de croissance contenant cet antibiotique. Plusieurs méthodes différentes peuvent être utilisées pour confirmer que les colonies cultivées dans les milieuis sélectifs sont des transformateurs (c’est-à-dire ont incorporé le plasmide). Par exemple, les plasmides peuvent être récupérés dans ces cellules à l’aide de méthodes de préparation au plasmide (10) et digérés pour confirmer la taille du plasmide. Alternativement, la colonie PCR peut être utilisée pour confirmer la présence du plasmide d’intérêt (11).
L’objectif de cette expérience est de préparer des cellules chimiquement compétentes E. coli DH5MD, en utilisant une adaptation de la procédure de chlorure de calcium (12), et de les transformer avec le plasmide pUC19 pour déterminer l’efficacité de la transformation. La souche E. coli DH5Estest une souche couramment utilisée dans des applications de biologie moléculaire. En raison de son génotype, en particulier le recA1 et endA1, cette souche permet une stabilité accrue de l’insertion et d’améliorer la qualité de l’ADN plasmide dans les préparations ultérieures. Étant donné que l’efficacité de transformation diminue avec l’augmentation de la taille de l’ADN, le plasmide pUC19 a été utilisé dans ce protocole en raison de sa petite taille (2686 bp) (voir https://www.mobitec.com/cms/products/bio/04_vector_sys/standard_cloning_vectors.html pour un carte vectorielle). pUC19 confère une résistance à l’ampicilline et donc, c’était l’antibiotique utilisé pour la sélection.